Lindsey R. FISCHER, Jonathan D. GLASS, Departments of Neurology, and Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, USA
Резюме
Данные, полученные при исследовании бокового амиотрофического склероза у животных и людей, все больше подтверждают предположение о том, что дистальная дегенерация аксонов при этом заболевании начинается на исключительно ранних стадиях. Это происходит гораздо раньше появления первых признаков гибели двигательных нейронов и возникновения отчетливой симптоматики. Причина аксональной дегенерации в настоящее время не выяснена. Возможно, что сочетание нескольких процессов приводит к развитию данного состояния. Вовлечение местных повреждающих факторов и резкое уменьшение трофической поддержки по причине изменения метаболизма окружающих тканей являются обязательными компонентами при развитии данной патологии. Совершенно понятно, что аксон по своей природе не является простым придатком тела нейрона, его дегенерация и гибель подчиняются тем же законам и правилам. Эти данные подтверждаются рядом исследований, в которых использовались объекты с искусственно созданным состоянием амиотрофического латерального склероза. При анализе указанных данных, связанных с методами защиты двигательных нейронов от любых травмирующих факторов, было установлено, что они не привели к сколько-нибудь значимым изменениям симптоматики или удлинению времени нормального функционирования исследуемых нейронов. Аналогичные данные были получены при попытке использовать технологии по защите непосредственно аксона, исследуемой клетки. В настоящей статье приводится развернутый анализ состояний ранней аксональной дегенерации при боковом амиотрофическом склерозе и обсуждаются возможные механизмы развития данного состояния с акцентированием внимания на процессы оксидативного стресса. Обсуждается первичное значение аксональной дегенерации как возможного механизма развития данного заболевания двигательных нейронов. Кроме того, предлагаются к рассмотрению факторы, предупреждающие развитие аксональной дегенерации, как важный аспект терапии данного состояния.
Ключевые слова
боковой амиотрофический склероз, аксональная дегенерация, нейромышечная связь, супероксиддисмутаза, оксидантный стресс
Введение
Аксональная дегенерация является одним из основных клинических и патологических факторов развития бокового амиотрофического склероза (БАС). Аксональная дегенерация в основном заканчивается дисфункцией клетки или ее гибелью, связанной с валлеровской или подобной дегенерацией. Сейчас опубликовано множество работ, касающихся экспериментальных данных, которые говорят, что любая защита тел мотонейронов от внешних воздействий имеет исключительно малую эффективность по отношению к скорости развития симптомов БАС. Фактически даже генетические вмешательства, которые, казалось, могли бы полностью изменить скорость развития патологического состояния в мотонейронах спинного мозга, до сих пор не в состоянии предотвратить или замедлить ослабление и гибель животных в экспериментальной модели заболевания мотонейронов. Этот кажущийся парадокс может быть объяснен тем, что невозможно уберечь тела нейронов от патологических процессов, которые уже развились в их аксонах. Прямым следствием повреждения аксонов является дегенеративный процесс, развивающийся в мышечной ткани. Указанные факты еще раз подчеркивают выдвинутую не так давно идею о том, что дегенерация аксонов двигательных нейронов не является прямым следствием дегенерации и гибели тела родительского нейрона. Кроме того, подчеркивается, что даже незначительных изменений в аксональном метаболизме будет достаточно для формирования клинической картины. Именно поэтому предупреждение аксональной дегенерации является важным с терапевтической точки зрения. Успех в достижении этой цели даст возможность улучшить терапию БАС и других нейродегенеративных заболеваний.
Аксональная дегенерация при раннем БАС
Что общего между аксональной дегенерацией двигательных нейронов при моделировании БАС и аналогичными состояниями у человека?
Моделирование на животных
Развитие патологического процесса, сопровождающегося нейрональной дегенерацией, в настоящее время широко исследуется на животных при моделировании данной патологии. Важную роль занимают также исследования, проводимые на пациентах с нейродистрофической патологией и с невропатией двигательных нейронов спинного мозга в частности. В подавляющем большинстве случаев развитие нейродистрофии является первичным процессом, обусловливающим недостаточность функции нейронов и проявление симптомов заболевания. При этом даже весьма выраженные парезы сопровождаются неожиданно легкими изменениями двигательных нейронов. Данные явления некоторые авторы объясняют изменениями в генетическом аппарате клеток, обусловливающими выраженные изменения их функций с легкими морфологическими отклонениями. Аксональная дегенерация, несомненно, является следствием аналогичных патологических процессов, приводящих к гибели нейрона, что, в свою очередь, вызывает денервацию соответствующей мышцы. Данные, касающиеся экспериментальных моделей, на которых производился анализ описанных ситуаций, приведены ниже.
Для анализа наследственного БАС наиболее широко используется моделирование на мутантных линиях мышей по Cu,Zn-супероксиддисмутазе. Высокая степень копирования мутантной SOD1 вызывает появление слабости на 80–90-й день и гибель животного на 130–140-й день. Оригинальные исследования показывают, что начало развития клинической картины у таких животных совпадает по времени с началом частичной гибели мотонейронов спинного мозга. При этом сопутствующие физиологические и патофизиологические исследования выявляют начало денервации мышц задних конечностей задолго до появления характерных симптомов заболевания. Для установления пространственно-временных особенностей развития процессов, приводящих к гибели мотонейрона, было проведено количественное исследование патологических процессов от конечной до начальной точки их развития. Значительные явления мышечной дегенерации появились на 47-й день, задолго до появления первых симптомов заболевания. На момент появления первых клинических признаков (80-й день исследования) две трети аксонов вентральных корешков уже были подвергнуты дегенерации. До момента развития ярких симптомов заболевания не встречалось ни одного случая выявления гибели тел мотонейронов. Аналогичные данные были представлены и другими лабораториями, которые работают с мутантными линиями мышей различных подтипов.
В серии работ использовались животные со специфической миссенс-мутацией, сопровождающейся дефектом синтеза тубулина в нейронах, что приводило к прогрессирующей моторной невропатии. У таких животных развитие слабости происходило в течение первых трех недель жизни, и гибель животных происходила на шестой неделе. Гибель мотонейронов или даже развитие дегенерации дистальной части аксонов не отмечались на фазе развития явных патологических проявлений. В конечной стадии развития данной патологии гибель мотонейронов встречалась не более чем в 40 % всех случаев. Электрофизиологические исследования показали некоторые изменения структуры потенциала действия в моторных нейронах, иннервирующих мускулатуру головы и задних конечностей животного. При этом прослеживались временные зависимости нарастания указанных изменений. Причем самые выраженные изменения наблюдались задолго до тринадцатого дня исследования. Очевидные патологические изменения, свидетельствующие о денервации указанных мышц, наблюдались после пятнадцатого дня исследования. Таким образом, можно сделать вывод об исключительно малой корреляции между выраженностью патологического процесса и клиническими проявлениями у данных животных.
Группа животных с моделированной спинальной мышечной атрофией, вызванной искусственным удалением седьмого экзона гена Smn, характеризовалась ранней развивающейся моторной деградацией и гибелью животного в возрасте около четырех недель. Патоморфологический анализ данных животных выявил, что было потеряно только около 30 % поясничных двигательных нейронов за тридцатидневный период. Кроме того, 49 % аксонов передних рогов сегментов L4–L5 подверглись деструкции в первые пятнадцать дней. Данный показатель вырос до 78 % к тридцатому дню. Денервация закономерно сопровождалась выраженным накоплением фосфорилированных нейрофиламентов в нейромышечных синапсах. Во всех случаях не было обнаружено явлений терминального прорастания, что может быть связано с сопровождающимся дефектом аксонального роста и пластичности. Анализируя спинальный уровень у данных животных, можно с уверенностью заключить, что дистальные аксональные дефекты были гораздо более выраженными по сравнению с деструкцией, происходящей в спинном мозге.
Во всех моделях большая часть двигательных нейронов была морфологически интактна в момент появления первых клинических признаков. Именно этот факт дает возможность предложить потенциально эффективное направление по предотвращению ухудшения состояния. При этом использование данных принципов у человека с БАС может помочь сохранить тела двигательных нейронов и при дальнейшей реиннервации улучшить состояние пациента.
БАС у людей
Аксональная дегенерация длительное время считалась единственной особенностью БАС у людей. При этом электромиографические исследования были основными в диагностике данных состояний. Такие особенности миограммы, как спонтанная фибрилляция и фастикуляция потенциалов, были наиболее важными в диагностике. Реиннервация при данном заболевании направлена на поддержание нормальной функции мышечной системы. Такое явление закономерно при БАС. Однако реиннервация не в состоянии адекватно компенсировать начавшуюся денервацию, которая и вызывает внезапную слабость соответствующих мышц. Денервацию традиционно приписывали формирующейся дисфункции и гибели мотонейронов. Помимо этого, реиннервация считалась следствием прорастания аксона моторного нейрона, лежащего в непосредственной близости от погибшего. Данные предположения не могли быть проверены на людях до момента смерти пациента с БАС.
Эти предположения поддерживаются еще и гипотезами о том, что БАС у людей начинается с дистальной дегенерации аксонов двигательных нейронов. Bradley с соавт. использовали количественные методики морфометрии для демонстрации проксимально-дистального градиента аксональной патологии в периферических нервах у людей с БАС. В нашем случае имелась редкая возможность оценить клинические, а затем патоморфологические данные у пациента с БАС, который умер во время оперативных вмешательств, не имеющих отношения к основному заболеванию. Клинический диагноз был установлен и прослежена клиника в течение не менее полугода с момента появления первых симптомов. Аутопсия подтвердила значительную денервацию и изменения в процессах реиннервации, которые были показаны миографически. Однако в данном случае не удалось выявить признаков, свидетельствующих об изменениях в мотонейронах спинного мозга.
В клинической практике исследователи использовали технологию определения числа моторных единиц для выявления количественных параметров процессов денервации и реиннервации. Данная технология позволила предположить, что уровень прогрессирования БАС у людей прямо зависит от денервации и потери функционирующих моторных единиц у пациента. Эти данные были проверены на линии мутантных мышей по супероксиддисмутазе-1. Технология подсчета моторных единиц использовалась и в этом случае. в ходе исследования получена важнейшая информация об уменьшении числа моторных единиц за несколько месяцев до начала формирования клинической картины. Для расширения спектра исследования использовалась методика порогового трекинга, которая позволяет выявить изменения аксональной возбудимости, что дало возможность получить также незаменимую информацию, касающуюся особенностей электрофизиологических изменений у пациентов с БАС. Явления резкого повышения натриевого тока и уменьшения калиевой проводимости наблюдались в двух популяциях пациентов. Особенностью этого состояния было значительное его усугубление в дистальных отделах аксонов. Несомненно, что ни одно из этих исследований не позволяет полностью раскрыть особенности течения БАС у людей. Однако в сочетании с данными, полученными на животных, можно с уверенностью говорить о том, что такое состояние является патологией дистальных отделов аксонов.
Последние исследования дали информацию еще об одном факторе, который может играть важную роль в формировании БАС. Именно информация о генетических изменениях при данной патологии заставляет все больше обращать на нее внимание. Говоря о генетических факторах, клиницисты должны иначе посмотреть на происхождение БАС. Из известных 43 мутаций, которые приводят к изменению двигательных нейронов, 14 причастны к формированию амиотрофического латерального склероза. Все мутации, имеющие место при БАС, влияют на такие принципиально важные моменты нейрональной биологии, как изменение цитоскелета нейронов, повреждение и трансформация медиаторных везикул, критические изменения аксонального транспорта. К роме того, важную роль в формировании патологического процесса играют нарушения ферментативных систем. Некоторые другие мутации вовлекают не только нейроны. В таких случаях причиной аномальной работы нервной клетки является дефект шванновских клеток. Роль этого типа глии сложно переоценить. Непосредственное участие в нейрональной передаче информации, в частности по моторным нейронам, осуществляется именно с помощью этих клеток. Все многообразие нарушений обмена клеток спинного мозга и окружающих тканей обусловливает постепенное развитие аксональной дегенерации, приводящей к моторной невропатии. Несмотря на все то обилие информации о явлениях, сопутствующих БАС, мы не можем сейчас более или менее четко сформировать стройную схему развития данной патологии. В наших силах проводить дополнительные исследования и подвергать более глубокому анализу уже полученные материалы.
Вовлечение сенсорных аксонов
Аксональная патология не ограничивается формированием БАС в эфферентном отделе ЦНС. Изменения в аксонах вовлекают и чувствительную часть нервной системы. Симптомы нарушения афферентных нейронов менее заметны и приводят к не настолько выраженным состояниям. Однако те изменения, которые встречаются у пациентов с БАС, также требуют коррекции. По усредненным данным, пациенты с БАС имеют снижение количества миелинизированных волокон в крупных нервных стволах до 70 % от исходного уровня. При этом количество данных волокон дорсальных корешков сегмента L5 уменьшается на 27 %. Изменения в телах нейронов, находящихся в околопозвоночных ганглиях на уровне 3–5-го поясничных сегментов, приводят к 54 % уменьшению их размеров. При анализе скорости проведения по сенсорным волокнам у пациентов с БАС выявляется значительное ее уменьшение в сравнении с контрольными группами пациентов. Исследования на мутантных животных выявили характерную особенность, которая заключалась в практически одновременном снижении количества моторных и сенсорных волокон при формировании БАС.
При анализе спинальной мышечной атрофии, вызванной прогрессированием БАС, выявлено, что имеет место снижение скорости проведения по смешанным нервам. При этом степень снижения колебалась от умеренной до выраженной. Одновременно проведенная биопсия выявила выраженное истончение нервных волокон. Гистологический анализ нервно-мышечных соединений у мутантных животных с БАС показал, что синаптические бляшки на соматической мускулатуре умеренно истончены, а данные, касающиеся анализа сенсорных волокон, свидетельствовали о практически полном исчезновении чувствительных нервных окончаний в верхних слоях кожи. В ходе анализа культуры нейронов, полученной от данных мышей, выявлялось, что их дендриты были значительно короче по сравнению с нормальными клетками. Аксональный холмик имел невыраженную структуру, количество т-РНК и м-РНК резко снижено в дистальных отделах аксонов. Обобщая полученные результаты, можно говорить об аналогичных процессах, происходящих в моторных и сенсорных нейронах, с учетом того, что в последних степень выраженности изменений несколько ниже.
Дегенерация сенсорных волокон при БАС не является прямым доказательством того, что именно двигательные нейроны являются местом развития всех патологических процессов при этом состоянии. Понимание причин того, что дегенеративные процессы развиваются в моторных аксонах при БАС с огромной скоростью и при этом сенсорные волокна остаются относительно интактными, дает ключ к дальнейшим изысканиям. Обнаружение причин и механизмов этих различий является первым шагом к лечению БАС. Больший размер двигательных нейронов, увеличенная метаболическая нагрузка, анатомическая организация, контакт с мышцами и другие отличия в структуре и функции двигательных нейронов рассматривались как причины формирования этой патологии. По нашим данным, прямые сравнения между чувствительностью моторных и сенсорных нейронов к действию таких потенциальных триггеров заболевания, как оксидативный стресс и экспрессия мутантной супероксиддисмутазы, проведены не были.
Механизмы аксональной дегенерации
При анализе литературы, касающейся влияния мутантной супероксиддисмутазы на дегенеративные процессы в аксонах, четкого описания этих процессов найдено не было. Кроме того, первичное место повреждения нервной системы также неизвестно. Отмирание части аксона может являться прямым следствием влияния травмирующего фактора на его дистальные отделы. Изучается и возможность перехода патологического процесса с тела нейрона на отростки. Системный или локальный принцип формирования БАС также рассматривался при анализе причин аксональной дегенерации. В данном разделе будут описаны механизмы дегенерации, присущие именно аксонам, а также будет рассмотрена возможность дистантного формирования БАС за счет использования мутантной супероксиддисмутазы как триггера аксональной дегенерации.
Аксональная дегенерация не зависит от гибели клеток
Очевидным является то, что аксональная дегенерация не является пассивным процессом. Она может быть лишь проявлением неизвестных нам процессов, часть из которых запрограммированна на гибель клетки. Например, удаление или уменьшение количества факторов роста нервной ткани из культуры клеток вызывает гибель тел клеток и их отростков. В опыте с мутантной линией мышей проводилось удаление проапоптотического гена, что вызвало сохранение двигательных нейронов у мышей с БАС. Однако денервация и дегенерация аксонов проявились вне зависимости от данной манипуляции. Все представленное выше подтверждает предположение о том, что аксональная дегенерация может происходить независимо от молекулярных процессов, контролирующих гибель клетки.
Концепция аксональной независимости была пересмотрена после открытия явления медленной валлеровской дегенерации, которая вызвана особым видом спонтанной мутации. Отличительной чертой в данном случае является удлиненная продолжительность жизни пораженных аксонов. Суть данного состояния сводится к тому, что мутантный ген обусловливает синтез нового химерного белка, имеющего протективное действие против химических и антигенных факторов, вызывающих аксональную дегенерацию. Несмотря на достаточно высокую эффективность данного белка, он все же не способен предотвратить дегенерацию или гибель клетки. Само существование такого агента, как химерный белок, имеющего уникальные свойства, свидетельствует о регулируемом процессе дегенерации клеток. Кроме этого, подтверждается гипотеза о различных механизмах клеточной и аксональной гибели. Эти данные позволяют предположить, что именно поиск в указанном направлении поможет найти инструмент для контроля за дегенеративными процессами в нервной ткани.
Эксперименты, проводящиеся на мутантных мышах с изменением в генотипе, подразумевающем наличие валлеровской дегенерации и прогрессирующей моторной невропатии, дали неоценимо важную информацию о разнонаправленности процессов сохранения тел и отростков нейронов. Мыши контрольной группы показали раннее развитие аксональной дегенерации в моторных нервах на сороковой день жизни. Введение дополнительной мутации, связанной с валлеровской дегенерацией, дало очень важный результат — неожиданно выраженное сохранение двигательных нейронов не только до сорокового дня, но и исключительно медленное развитие дегенеративных изменений, продолжавшихся до двух месяцев. Кроме этого, получены данные о том, что именно состояние аксонов прямо влияет на сохранность тела нейрона. Аналогичным образом проводилось тестирование групп мышей с мутацией по супероксиддисмутазе. Однако, к сожалению, при таком подходе было выявлено лишь незначительное изменение прогрессирования дегенерации, причем замедление этого процесса отмечалось только на первых этапах работы. Дополнительное исследование, проведенное с учетом всех поставленных задач, дало возможность достоверно предполагать, что протективные свойства атипичного белка, образующегося при валлеровской дегенерации, действенны первые две недели развития процесса. В последующие несколько месяцев сам химерный белок начинает работать как фактор, усугубляющий течение аксональной дегенерации. С учетом этих особенностей исследуемого белка был сделан вывод о возможности частичной приостановки подобного процесса дегенеративных изменений аксонов мотонейронов, который стремительно наблюдается при прогрессирующей моторной невропатии. К сожалению, использование этого же белка не имеет смысла в лечении хронической или вялотекущей дегенерации нервной ткани. Механизм действия протективного фактора, образующегося при валлеровской дегенерации, на сегодня не известен, однако его терапевтические эффекты могут найти применение в лечении огромного спектра патологических состояний. Определение гена или совокупности генов, ответственных за синтез протективного фактора при медленно прогрессирующей валлеровской дегенерации, не привело к успеху. Сейчас возможно его получение только при изменении достаточно крупного участка ДНК животного.
Роль кальпаина в развитии аксональной дегенерации
Исследования молекулярного механизма аксональной дегенерации поддерживают идею о неапоптотической программе гибели, развивающейся в аксоне. Практически при всех состояниях, ведущих к дегенеративным изменениям нервной клетки, первичными проявлениями являются замедление аксонального транспорта, митохондриальная дисфункция, увеличение концентрации кальция в клетке, что в конечном итоге приводит к активации системы кальпаинов. Кальпаины представляют собой группу нейтральных протеаз, активируемых кальцием. Их роль в формировании различных патологических процессов достаточно полно описана. Моделирование патологических состояний, проводившееся на животных и отдельных клетках, четко демонстрирует дегенерацию белков цитоскелета и мембраны клетки, вызванную кальпаином. При моделировании ишемических, травматических, токсических и других состояний результаты в отношении роли кальпаиновой системы были аналогичными. Ингибирование кальпаинов при периферической невропатии приводит к заметному снижению травмирующего влияния этих ферментов на клеточные элементы как в эксперименте на животном, так и на культуре клеток. Ингибирование кальпаинов также приводит к сохранению цитоскелета в нервно-мышечном соединении в модели острой аутоиммунной нервно-мышечной дегенерации. Полученные данные о роли кальпаинов при развитии нейрональной дегенерации подтверждаются многими авторами. Кроме того, в значительной части случаев ингибиторы кальпаинов предупреждают или замедляют развитие патологических процессов в нервной клетке. Однако принципиальным отличием данных работ является то, что акцент делается именно на гибель тела нейрона. Изменения, проходившие в отростках этих клеток, во внимание не принимались. Как бы там ни было, применение ингибиторов кальпаинов при лечении аксональной дегенерации может действительно оказаться тем фактором, который будет играть решающую роль.
Чем обусловлена аксональная дегенерация при амиотрофическом латеральном склерозе?
Причина или несколько причин аксональной дегенерации при БАС у мутантной линии мышей по супероксиддисмутазе не выяснена. Некоторые ученые рассматривают ее токсическое действие на клеточные элементы. Механизмы токсического влияния включают в себя агрегацию мутантных протеинов, поломку митохондриальных систем и токсическое действие глутамата. Все перечисленные факторы в отдельности или ассоциированно с факторами, вызывающими запуск апоптоза, приводят к характерным изменениям в клетке. Каждый из этих процессов достаточно хорошо изучен в отдельности и применительно к спинальным мотонейронам. Однако комплексный анализ с учетом особенностей процессов, протекающих в дистальных отделах аксонов, не проводился.
Дефекты аксонального транспорта
Процесс ухудшения работы быстрого антероградного аксонального транспорта у мутантных мышей по супероксиддисмутазе и у людей с БАС достаточно глубоко изучен. Заметное снижение скорости антероградного транспорта по аксонам с накоплением нейрофиламентов в проксимальной части нейрона описано у мутантных мышей и людей с БАС. При этом в спинном мозге признаки такого явления появляются в среднем за полгода до первых клинических проявлений заболевания. Замедление ретроградного транспорта также замечено в моторных нейронах мутантных мышей с 13-го дня эмбрионального развития. Такие явления, как изменение скорости транспорта веществ по отросткам нейрона, могут быть прямой причиной нарушения ее функции, а также следствием и симптомом такого нарушения. Выяснение истинной роли изменения скорости аксонального транспорта в аксональной дистрофии еще предстоит выяснить. Недавно были идентифицированы мутации, проявляющиеся в изменении комплекса белковых молекул с динеин/динактином, которые являются характерными именно для пораженных двигательных нейронов у мышей и людей. Нормализуя аксональный транспорт, достаточно часто удавалось пролонгировать жизнедеятельность клетки. Указанные данные свидетельствуют о том, что активность и состоятельность аксонального транспорта является еще одним критическим звеном в цепи патогенеза аксонального амиотрофического склероза.
Оксидативный стресс
При описании БАС и некоторых других нейродегенеративных процессов традиционно указывается на роль оксидативного повреждения как фактора, обусловливающего старение клетки и/или организма. Абсолютно очевидно, что в трупных тканях оксидативный процесс в отношении белков, липидов и ДНК при БАС выражен исключительно сильно. Однако какова роль перекисного окисления в формировании ранних стадий заболевания, на сегодня не выяснено. Аналогично выявлена роль оксидативного повреждения клеток у линии мутантных мышей. Абсолютно очевидно повышение интенсивности перекисного окисления в клетках спинного мозга после месячного периода развития симптомов болезни. Информация, касающаяся интенсивности оксидативного повреждения клеток в период, предшествующий появлению яркой симптоматики, является весьма скудной. Исходя из того, что абсолютное большинство клеточных элементов, белков, частей цитоскелета и др. попадают в дистальные части аксонов за счет аксонального транспорта, логично предположить, что именно дистальная часть отростка нейрона содержит наиболее «старые» клеточные единицы. Эти части клетки в большей мере подвергались травмирующему действию перекисного окисления, вследствие чего резервы и «запас прочности» у таких элементов снижены, а это значит, что дегенерация и дисфункция этих компонентов наиболее вероятна.
Несомненно то, что нейромышечные соединения могут быть уязвимы для повреждения окислительными процессами, активными у животных с мутацией по супероксиддисмутазе. До тех пор, пока у них не развивается дистрофия двигательных нейронов, значительных симптомов заболевания не наблюдается. И только после развития выраженной моторной аксонопатии происходит резкое ускорение процессов старения и появление мышечной атрофии. Миографические исследования показывают спонтанную активность пораженных мышц, что свидетельствует о начавшейся денервации. Среди миографических симптомов данного состояния встречаются фибрилляции и сложные комплексные разряды. Анализируя материалы гистологического исследования, можно сказать о начинающейся денервации дистальных отделов конечностей на втором месяце заболевания, и к концу первого полугодия формируется комбинация атрофии мышечных волокон и их гипертрофии. Кроме того, среди характерных признаков наблюдается группирование мышечных волокон, что указывает на хроническую моторную невропатию.
Предложена гипотеза о том, что клиническое и патофизиологическое сходство между разными группами мутантных мышей, отличавшихся только степенью выраженности экспрессии генов, кодирующих супероксиддисмутазу, заключается лишь в выраженности симптомов. Главный смысл состоит в том, что оксидативный стресс играет основную роль в аксональной дегенерации, наблюдающейся у животных данных групп. Интенсивность оксидативного стресса может быть достаточной для формирования или запуска процессов аксональной патологии. Например, увеличение размера митохондрий в нейромышечном соединении становится заметным на 25-й день развития патологического процесса в нервной ткани. Это указывает на значительное повреждение митохондриальной структуры. Оксидативное повреждение митохондрий нарушает динамику электронов, тем самым угнетая синтез АТФ, вплоть до его остановки. Это является еще одним фактором, замедляющим аксональный энергозависимый транспорт. Недостаточность энергетического снабжения транспортных систем нейрона приводит к остановке двустороннего обмена между телом и отростками нейрона. Замедление транспорта является нормальным проявлением старения нервных клеток. Как было описано, включается «порочный круг», связанный с увеличением оксидативного влияния на органеллы. АТФ также является жизненно важным компонентом для работы Na+/K+-АТФазы. Дисфункция этого фермента влечет за собой накопление натрия в аксоне. Повышение концентрации данного иона приводит к реверсу натрий-кальциевого транспорта с входом и накоплением кальция в аксоне. Как было описано выше, кальций является прямым активатором кальпаиновой системы, что резко ускоряет дегенеративные процессы в аксональных структурах. Помимо повреждения митохондрий, оксидативный стресс влияет на жизнедеятельность других клеточных компонентов, содержащихся в аксоне. Накопление гиперфосфорилированных нейрофиламентов в дистальных отделах аксонов — типичное явление для пациентов с БАС. Этот феномен является типичным для состояний, связанных с повышением активности Cdk5, встречающимся при оксидативном стрессе. В дополнение к огромному количеству эффектов, к которым приводит оксидативный стресс, свободные радикалы резко снижают эффективность выделения медиаторов из пресинапса моторных терминалей. Эксперименты, модулирующие аналогичные состояния за счет применения ботулина, имели стандартный и закономерный результат, заключающийся в повреждении дистальных отделов моторных аксонов, и даже приводили к гибели двигательных нейронов.
Влияние свободных радикалов на компоненты клеток невероятно многообразно. Оно проявляется в изменении свойств белковых и липидных молекул, что вызывает различные дегенеративные процессы. Перекисное окисление мутантной супероксиддисмутазы, как известно, приводит к прекращению функционирования этого фермента. Действие перекисного окисления на убиквитин-карбокси-терминальную гидролазу аналогично ее полной элиминации из клетки. Вследствие этого возникает так называемая грациллярная аксональная дистрофия. Обращает на себя внимание то, что нейрофиламенты исключительно чувствительны к оксидативному повреждению. Экспозиция очищенных нейрофиламентов в супероксиддисмутазе и пероксиде водорода вызывает дозозависимую агрегацию указанных белков. Агрегированные нейрофиламенты за счет действия любого из возможных факторов перекисного окисления, в частности аскорбата трихлористого железа, становятся более чувствительными к действию кальпаинов. Это, в свою очередь, приводит к заметному увеличению скорости дегенерации. В клетках нейробластомы человека высокие дозы аскорбата вызывают дегенерацию нейрофиламентов, имеющую дозозависимый эффект, что впоследствии приводит к гибели клетки. В отличие от нейрофиламентов тубулин и актин не подвергаются агрегации в такой модели перекисного окисления. Это означает, что нейрофиламенты имеют практически уникальную чувствительность к оксидативному стрессу. Такая их особенность может быть связана с наличием специфических локусов, сопряженных с лизином и пролином. Эти части белковой молекулы теоретически становятся мишенью для модификации свободными радикалами, что приводит к альтерации и изменению вторичной структуры белка. В модели цитоскелета свободные радикалы приводят к таким критическим изменениям белковых структур, которые вызывают его деструкцию. Данные состояния могут быть предупреждены или уменьшены введением витаминов Е и С.
Особенности проявления аксональной дегенерации указывают на исключительную важность оксидативных процессов в ее формировании. Так, денервация моторных соединений у больных мышей происходит по определенной закономерности. Первыми дегенерируют нейроны и их отростки, имеющие малый и средний уровень резервов. Медленные моторные единицы в меньшей степени подвержены дегенерации. Кроме того, в них быстрее происходит регенерация и восстановление отростков. Общая картина развития нейромышечных заболеваний отражает естественные процессы старения. Единственное отличие — скорость развития этих состояний. Интересным фактом является то, что наиболее уязвимы для оксидативного стресса клетки, в которых уровень окислительного фосфорилирования не слишком высок. Обычно именно в таких клетках количество элементов антиоксидантной системы гораздо ниже. Соответственно склонность к дегенеративным процессам нервной ткани может быть диагностирована по количеству и активности антиоксидантов в данном организме. Еще одним интересным наблюдением является то, что клетки, находящиеся в непосредственной близости к поврежденным элементам, увеличиваются в объеме. Также проявляется увеличенное количество мышечных элементов, компенсаторно иннервируемое ими. Предполагаемой причиной развития дисбаланса между процессами перекисного окисления и антиоксидантной системой может быть слишком быстрая экспрессия мутантного гена, кодирующего синтез супероксиддисмутазы, сдвигающее равновесие систем в сторону перекисного окисления и оксидативного стресса.
Многочисленные антиоксиданты тестировались при выполнении данного исследования. Некоторые из них проявили значительную антиоксидантную активность. Как результат удалось пролонгировать выживаемость мутантных мышей до двух раз относительно контрольной группы. При этом назначение антиоксидантов проводилось в период скрытого, бессимптомного течения заболевания. Тестирование активности антиоксидантных систем организма на пациентах, по результатам метаанализа, не принесло статистически значимого результата. Вследствие того, что измерение степени оксидативного стресса на сегодняшний день не представляется возможным, не удается установить причину отсутствия результата назначение антиоксидантной терапии пациентам. Так, неэффективность лечения может быть связана и с повышением степени окислительных реакций, и с недостаточной эффективностью предлагаемой терапии. Даже если оксидативный процесс играет одну из основных ролей в запуске цепочки реакций, приводящих к аксональной дегенерации, мы не можем с помощью медикаментов предотвратить этот процесс. Таким образом, для разработки эффективной методики лечения оксидативных повреждений при БАС нам необходимо еще более глубокое исследование цепочки патологических процессов, приводящих к этим изменениям.
Роль других клеток
В предшествующих разделах обсуждались явления в нейроне и его частях, которые могут играть важную роль в аксональной дегенерации. Несмотря на несомненно важную роль процессов, происходящих непосредственно в самой клетке, однозначным является то, что дегенеративные изменения при БАС не проходят изолированно только в нейронах. Реакции глиальных клеток также являются достаточно важным прогностическим критерием и признаком степени изменений, уже произошедших в ЦНС. Менее известны феномены, происходящие в клетках, находящихся в непосредственной близости от дистальных участков аксонов. К таким клеточным элементам можно отнести шванновские, мышечные и даже иммунные клетки.
Шванновские клетки являются наиболее крупными глиальными клетками, находящимися в ЦНС. Они имеют контакт с аксонами нервных клеток на огромной площади. Эти участки превосходят места контактов нейронов с астроцитами в несколько десятков раз. Не следует упускать из виду то, что эти клетки являются еще и значительными источниками трофической поддержки и физической защиты нейронов. Таким образом, можно считать, что именно шванновские клетки и нейроглиальные союзы являются наиболее подходящими объектами исследования для выяснения природы гибели нейронов. Мутантная супероксиддисмутаза обнаруживается в шванновских клетках мышей мутантной линии в первые дни постнатального периода. В терминальных шванновских клетках в острый период дегенерации нейронов наблюдается резкое повышение аксонального фактора семафорина-3А. Этот факт означает попытки шванновских клеток противостоять травмирующим факторам за счет синтеза ингибиторов перекисного окисления, проявляющих свойства стимуляторов регенерации. Таким образом, эти клетки глии на некоторое время задерживают начинающуюся дисфункцию нейромышечного соединения и замедляют развитие симптомов БАС.
При аналогичном анализе скелетной мускулатуры в миоцитах также обнаруживается высокий уровень мутантной супероксиддисмутазы, которая аналогичным образом действует на нейромышечное соединение. Некоторые авторы считают, что роль миоцитов заключается в запуске патологических реакций, направленных на дегенерацию нейромышечного соединения. Именно мышечная часть этого образования является причиной данной патологии. Однако достоверных данных, подтверждающих эту теорию, получено не было. Увеличение синтеза фактора роста и ветвления дендритов у пациентов с БАС и у животных мутантных линий вызвало создание гипотезы о возможной роли миоцитов в ингибировании регенеративных процессов дистальных отделов аксонов. В подтверждение этой гипотезы можно привести следующие факты: при ингибировании рядом ферментов указанных факторов роста дендритов увеличение продолжительности жизни мутантных мышей составило порядка 25 дней.
Последние несколько исследований были направлены на выяснение действительной роли миоцитов в формировании дегенерации нейронов у мутантных мышей. Отделение миоцитов из нейромышечного соединения не показало достоверных изменений продолжительности жизни животных мутантных линий. Кроме того, результатом данной работы было лишь незначительное снижение мутантных белков в исследуемых нейронах. Необходимо проведение дополнительных серий экспериментов, которые уточнят роль мышечных клеток в патологии двигательных нейронов. Кроме того, необходимо определить дальнейшие направления исследования для выяснения возможности использования компонентов миоцитов в лечении БАС.
Заключение
В результате масштабных исследований получены очевидные данные, касающиеся патогенеза нейрональной дегенерации. Неоспорим тот факт, что процессы аксональной дегенерации и изменения, происходящие в теле нейрона, наблюдаются независимо друг от друга. Поэтому чрезвычайно важно помнить, что мероприятия, направленные на поддержание жизнедеятельности тела нейрона, не повлияют на клинические проявления БАС и продолжительность жизни. С другой стороны, были выявлены критические зоны развивающейся патологии. С точки зрения авторов именно на них должна быть направлена терапия при дегенеративных состояниях нейронов. Механизм развития аксональной дегенерации до настоящего времени остается неизвестным. Однако с уверенностью можно сказать, что одной из важных фаз развития данных состояний является оксидативный стресс. Множество связанных с ним факторов могут внезапно спровоцировать резкое увеличение реакций перекисного окисления, что вызовет процессы ускоренного старения и, как следствие, дегенерации нервной ткани. В дополнение можно сказать, что роль шванновских клеток в развитии дегенеративных процессов нельзя недооценивать. Это связано именно со свойством данных клеток стимулировать регенеративные процессы в нейронах. Дальнейшие исследования, несомненно, дадут новые факты, которые позволят с иной стороны подойти к рассмотрению патогенеза дегенеративных заболеваний нервной ткани, в частности, к амиотрофическому латеральному склерозу.
Литература
1. Glass J.D. Wallerian degeneration as a window to peripheral neuropathy // J. Neurol. Sci. 2004; 220: 123-124.
2. Gould T.W., Buss R.R., Vinsant S., Prevette D. et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS // J. Neurosci. 2006; 26: 8774-8786.
3. Kostic V., Jackson-Lewis V., de Bilbao F., Dubois-Dauphin M., Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis // Science 1997; 277: 559-562.
4. Li M., Ona V.O., Guegan C., Chen M., Jackson-Lewis V. et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model // Science 2000; 288: 335-339.
5. Sagot Y., Dubois-Dauphin M., Tan S.A., de Bilbao F. et al. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease // J. Neurosci. 1995; 15: 7727-7733.
6. Klivenyi P., Ferrante R.J., Matthews R.T., Bogdanov M.B. et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis // Nat. Med. 1999; 5: 347-350.
7. Chiu A.Y., Zhai P., Dal Canto M.C., Peters T.M. et al. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis // Mol. Cell. Neurosci. 1995; 6: 349-362.
8. Kennel P.F., Finiels F., Revah F., Mallet J. Neuromuscular function impairment is not caused by motor neuron loss in PALS mice: an electromyographic study // Neuroreport. 1996; 7: 1427-1431.
9. Frey D., Schneider C., Xu L., Borg J., Spooren W., Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases // J. Neurosci. 2000; 20: 2534-2542.
10. Fischer L.R., Culver D.G., Tennant P., Davis A.A. et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man // Exp. Neurol. 2004; 185: 232-240.
11. Pun S., Santos A.F., Saxena S., Xu L., Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF // Nat. Neurosci. 2006; 9: 408-419.
12. Schaefer A.M., Sanes J.R., Lichtman J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis // J. Comp. Neurol. 2005; 490: 209-219.
13. Bommel H., Xie G., Rossoll W., Wiese S., Jablonka S., Boehm T., Sendtner M. Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease // J. Cell. Biol. 2002; 159: 563-569.
14. Martin N., Jaubert J., Gounon P., Salido E., Haase G., Szatanik M., Guenet J.L. A missense mutation in Tbce causes progressive motor neuronopathy in mice // Nat. Genet. 2002; 32: 443-447.
15. Holtmann B., Zielasek J., Tokya K.V., Sendtner M. Comparative analysis of motoneuron loss and functional deficits in PMN mice: implications for human motoneuron disease // J. Neurol. Sci. 1999; 169: 140-147.
16. Sendtner M., Schmalbruch H., Stockli K.A. et al. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy // Nature 1992; 358: 502-504.
17. Cifuentes-Dias C., Nicole S., Velasco M.E. et al. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model // Hum. Mol. Genet. 2002; 11: 1439-1447.
18. Hansen S., Ballantyne J.P. A quantitative electrophysiological study of motor neuron disease // J. Neurol. Neurosurg. Psychiatry 1978; 41: 773-783.
19. Bradley W.G., Good P., Rasool C.G., Adelman L.S. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis // Ann. Neurol. 1983; 14: 267-277.
20. Shefner J.M. Motor unit number estimation in human neurological diseases and animal models // Clin. Neurophysiol. 2001; 112: 955-964.
21. Armon C., Brandstater M.E. Motor unit number estimate-based rates of progression of ALS predict patient survival // Muscle Nerve 1999; 22: 1571-1575.
22. Yuen E.C., Olney R.K. Longitudinal study of fiber density and motor unit number estimate in patients with amyotrophic lateral sclerosis // Neurology 1997; 49: 573-578.
23. Shefner J.M., Cudkowicz M., Brown R.H. Jr. Motor unit number estimation predicts disease onset and survival in a transgenic mouse model of amyotrophic lateral sclerosis // Muscle Nerve 2006; 34: 603-607.
24.Aggarwal A., Nicholson G. Detection of pre-clinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation // J. Neurol. Neurosurg. Psychiatry 2002; 73: 199-201.
25. Kanai K., Kuwabara S., Misawa S., Tamura N. et al. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage // Brain 2006; 129: 953-962.
26.Vucic S., Kiernan M.C. Axonal excitability properties in amyotrophic lateral sclerosis // Clin. Neurophysiol. 2006; 117: 1458-1466.
27. Nakata M., Kuwabara S., Kanai K., Misawa S. et al. Distal excitability changes in motor axons in amyotrophic lateral sclerosis // Clin. Neurophysiol. 2006; 117: 1444-1448.
28. Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics // Nat. Rev. Neurosci. 2006; 7: 710-723.
29. Heads T., Pollock M., Robertson A., Sutherland W.H.F., Allpress S. Sensory nerve pathology in amyotrophic lateral sclerosis // Acta Neuropathol. 1991; 82: 316-320.
30. Kawamura Y., Dyck P.J., Shimono M., Okazaki H., Tateishi J., Doi H. Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis // J. Neuropathol. Exp. Neurol. 1981; 40: 667-675.
31. Shefner J.M., Tyler R., Krarup C. Abnormalities in the sensory action potential in patients with amyotrophic lateral sclerosis // Muscle Nerve 1991; 14: 1242-1246.
32. Theys P.A., Peelers E., Robberecht W. Evolution of motor and sensory deficits in amyotrophic lateral sclerosis estimated by neurophysiological techniques // J. Neurol. 1999; 246: 438-442.
33. Fischer L.R., Culver D., Davis A.A., Tennant P. et al. The WldS gene modestly prolongs survival in the SOD1-G93A fALS mouse // Neurobiol. Dis. 2005; 19: 293-300.
34. Anagnostou E., Miller S.P., Guiot M.C., Karpati G. et al. Type I spinal muscular atrophy can mimic sensory-motor axonal neuropathy // J. Child Neurol. 2005; 20: 147-150.
35. Rudnik-Schoneborn S., Goebel H.H., Schlote W. et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy // Neurology 2003; 60: 983-987.
36. Jablonka S., Karle K., Sandner B., Andreassi C., von Au K., Sendtner M. Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy // Human Mol. Genet. 2006; 15: 511-518.
37. Finn J.T., Weil M., Archer F., Siman R., Srinivasan A., Raff M.C. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases // J. Neurosci. 2000; 20: 1333-1341.
38. Sagot Y., Vejsada R., Kato A. Clinical and molecular aspects of motoneurone diseases: animal models, neurotrophic factors and Bcl-2 oncoprotein // Trends Pharmacol. Sci. 1997; 18: 330-337.
39. Lunn E.R., Perry V.H., Brown M.C., Rosen H., Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve // Eur. J. Neurosci. 1989; 1: 27-33.
40. Conforti L., Tarlton A., Mack T.G.A., Mi W. et al. A Ufd2/D4Colele chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wld s ) mouse // Proc. Natl. Acad. Sci. USA 2000; 97: 11377-11382.
41. Wang M.S., Davis A.A., Culver D.G., Glass J.D. WldS mice are resistant to paclitaxel (taxol) neuropathy // Ann. Neurol. 2002; 52: 442-447.
42. Wang M.S., Fang G., Culver D.G., Davis A.A., Rich M.M., Glass J.D. The Wld s protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy // Ann. Neurol. 2001; 50: 773-779.
43. Wang M.S., Wu Y., Culver D.G., Glass J.D. The gene for slow Wallerian degeneration (Wld s ) is also protective against vincristine neuropathy // Neurobiol. Dis. 2001; 8: 155-161.
44.Ferri A., Sanes J.R., Coleman M.P., Cunningham J.M., Kato A.C. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease // Curr. Biol. 2003; 13: 669-673.
45. Samsam M., Mi W., Wessig C., Zielasek J. et al. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy // J. Neurosci. 2003; 23: 2833-2839.
46. Deckwerth T.L., Johnson J., Eugene M. Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) // Dev. Biol. 1994; 165: 63-72.
47. Adalbert R., Nogradi A., Szabo A., Coleman M.P. The slow Wallerian degeneration gene in vivo protects motor axons but not their cell bodies after avulsion and neonatal axotomy // Eur. J. Neurosci. 2006; 24: 2163-2168.
48. Coleman M. Axon degeneration mechanisms: commonality amid diversity // Nat. Rev. Neurosci. 2005; 6: 889-898.
49. Raff M.C., Whitmore A.V., Finn J.T. Axonal self-destruction and neurodegeneration // Science 2002; 296: 868-871.
50. Coleman M.P., Perry V.H. Axon pathology in neurological disease: a neglected therapeutic target // Trends Neurosci. 2002; 25: 532-537.
51. Vande Velde C., Garcia M.L., Yin X., Trapp B.D., Cleveland D.W. The neuroprotective factor Wlds does not attenuate mutant SODl-mediated motor neuron disease // Neuromolecular Med. 2004; 5: 193-203.
52. Crawford T.O., Hsieh S.T., Schryer B.L., Glass J.D. Prolonged axonal survival in transected nerves of C57BL/Ola mice is independent of age // J. Neurocytol. 1995; 24: 333-340.
53. Gillingwater T.H., Thomson D., Mack T.G.A. et al. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld s mutant and Ube4b/Nmnat transgenic mice // J. Physiol. 2002; 543: 739-755.
54.Araki T., Sasaki Y., Millbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration // Science 2004; 305: 1010-1013.
55. Wang J., Zhai Q., Chen Y., Lin E., Gu W., McBurney M.W., He Z. A local mechanism mediates NAD-dependent protection of axon degeneration // J. Cell. Biol. 2005; 170: 349-355.
56. Conforti L., Fang G., Beirowski B., Wang M.S. et al. NAD(+) and axon degeneration revisited: Nmnatl cannot substitute for Wld(S) to delay Wallerian degeneration // Cell Death Differ. 2007; 14: 116-127.
57. Bartus R., Elliott P., Hayward N. Calpain as a novel target for treating acute neurodegenerative disorders // Neurol. Res. 1995; 17: 249-258.
58. Wang M.S., Wu Y., Culver D., Glass J.D. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincris-tine neuropathy // J. Neuropathol. Exp. Neurol. 2000; 59: 599-606.
59. Wang M.S., Davis A.A., Culver D.G., Wang Q., Powers J.C., Glass J.D. Calpain inhibition protects against Taxol-induced sensory neuropathy // Brain 2004; 127: 671-679.
60. O’Hanlon G.M., Humphreys P.D., Goldman R.S. et al. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury // Brain 2003; 126: 2497-2509.
61. Adamec E., Mohan P., Vonsattel J.P., Nixon R.A. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2 // Acta Neuropathol. 2002; 104: 92-104.
62. Nixon R.A., Saito K.I., Grynspan F., Griffin W.R. et al. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease // Ann. NY Acad. Sci. 1994; 747: 77-91.
63. Kieran D., Greensmith L. Inhibition of cal-pains, by treatment with leupeptin, improves motoneuron survival and muscle function in models of motoneuron degeneration // Neuroscience 2004; 125: 427-439.
64. Li J., Nixon R.A., Messer A., Berman S., Bursztajn S. Altered gene expression for calpain/calpastatin system in motor neuron degeneration (Mnd) mutant mouse brain and spinal cord // Brain Res. Mol. Brain Res. 1998; 53: 174-186.
65. Brown J., Robert H. Superoxide dismutase in familial amyotrophic lateral sclerosis: models for gain of function // Curr. Opin. Neurobiol. 1995; 5: 841-846.
66. Cleveland D.W., Rothstein J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS // Nat. Rev. Neurosci. 2001; 2: 806-819.
67. Boillee S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors // Neuron 2006; 52: 39-59.
68. Williamson T.L., Cleveland D.W. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons // Nat. Neurosci. 1999; 2: 50-56.
69. Zhang B., Tu P.H., Abtahian F., Trojanowski J.Q., Lee V.M.Y. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation // J. Cell. Biol. 1997; 139: 1307-1315.
70. Warita H., Itoyama Y., Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene // Brain Res. 1999; 819: 120-131.
71. Sasaki S., Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis // Neurology 1996; 47: 535-540.
72. Murakami T., Nagano I., Hayashi T., Manabe Y. et al. Impaired retrograde axonal transport of adenovirus-mediated E. coli LacZ gene in the mice carrying mutant SOD1 gene // Neurosci. Lett. 2001; 308: 149-152.
73. Kieran D., Hafezparast M., Bohnert S. et al. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice // J. Cell. Biol. 2005; 169: 561-567.
74. Hafezparast M., Klocke R., Ruhrberg C. et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport // Science 2003; 300: 808-812.
75. LaMonte B., Wallace K.E., Holloway B.A. et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive deterioration // Neuron 2002; 34: 715-727.
76. Puls I., Jonnakuty C., LaMonte B., Holzbaur E.L.F. et al. Mutant dynactin in motor neuron disease // Nat. Genet. 2003; 33: 455-456.
77. Balaban R.S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging // Cell 2005; 120: 483-495.
78. Barber S.C., Mead R.J., Shaw P.J. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target // Biochim. Biophys. Acta 2006; 1762: 1051-1067.
79. Hall E.D., Andrus P.K., Oostveen J.A., Fleck T.J., Gurney M.E. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS // J. Neurosci. Res. 1998; 53: 66-77.
80. Shefner J.M., Reaume A.G., Flood D.G. et al. Mice lacking cytosolic copper/zinc superoxide dismutase display a distinctive motor axonopathy // Neurology 1999; 53: 1239-1246.
81. Muller P.L., Song W., Liu Y. et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy // Free Radic. Biol. Med. 2006; 40: 1993-2004.
82. Flood D.G., Reaume A.G., Gruner J.A., Hoffman E.K. et al. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions // Am. J. Pathol. 1999; 155: 663-672.
83. Echtay K.S., Roussel D., St-Pierre J., Jekabsons M.B. et al. Superoxide activates mitochondrial uncoupling proteins // Nature 2002; 415: 96-99.
84. Fernandez H.L., Hodges-Savola C.A. Axoplasmic transport of calcitonin gene-related peptide in rat peripheral nerve as a function.of age // Neurochem. Res. 1994; 19: 1369-1377.
85.Frolkis V.V., Tanin S.A., Gorban Y.N. Age-related changes in axonal transport // Exp. Gerontol. 1997; 32: 441-450.
86. Shea T.B., Zheng Y.L., Ortiz D., Pant H.C. Cyclin-dependent kinase 5 increases perikaryal neurofilament phosphorylation and inhibits neurofilament axonal transport in response to oxidative stress // J. Neurosci. Res. 2004; 76: 795-800.
87. Giniatullin A.R., Darios F., Shakirzyanova A., Davletov B., Giniatullin R. SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release // J. Neurochem. 2006; 98: 1789-1797.
88. Berliocchi L., Fava E., Leist M., Horvat V., Dinsdale D., Read D., Nicotera P. Botulinum neurotoxin C initiates two different programs for neurite degeneration and neuronal apoptosis // J. Cell Biol. 2005; 168: 607-618.
89. Hodgson E.K., Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme // Biochemistry 1975; 14: 5294-5299.
90. Poon H.P., Hensley K., Thongboonkerd V., Merchant M.L. et al. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice — a model of familial amyotrophic lateral sclerosis // Free Radic. Biol. Med. 2005; 39: 453-462.
91. Saigoh K., Wang Y.L., Suh J.G., Yamanishi T., Sakai Y. et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice // Nat. Genet. 1999; 23: 47-51.
92. Kim N.H., Jeong M.S., Choi S.Y., Hoon Kang J. Oxidative modification of neurofilament-L by the Cu,Zn-superoxide dismutase and hydrogen peroxide system // Biochimie 2004; 86: 553-559.
93. Troncoso J.C., Costello A.C., Kim J.H., Johnson G.V. Metal-catalyzed oxidation of bovine neurofilaments in vitro // Free Radic. Biol. Med. 1995; 18: 891-899.
94. Cookson M.R., Thatcher N.M., Ince P.G., Shaw P.J. Selective loss of neurofilament proteins after exposure of differentiated human IMR-32 neuroblastoma cells to oxidative stress // Brain Res. 1996; 738: 162-166.
95. Gelinas S., Chapados C., Beauregard M., Gosselin I., Martinoli M.G. Effect of oxidative stress on stability and structure of neurofilament proteins // Biochem. Cell Biol. 2000; 78: 667-674.
96. Counterman A.E., D’Onofrio T.G., Andrews A.M., Weiss P.S. A physical model of axonal damage due to oxidative stress // Proc. Natl. Acad. Sci. USA 2005; 103: 5262-5266.
97. Gordon T., Hegedus J., Tarn S.L. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease // Neurol. Res. 2004; 26: 174-185.
98. Luff A.R. Age-associated changes in the innervation of muscle fibers and changes in the mechanical properties of motor units // Ann. NY Acad. Sci. 1998; 854: 92-101.
99. Hollander J., Fiebig R., Gore M., Bejma J., Ookawara T., Ohno H., Ji L.L. Superoxide dismutase gene expression in skeletal muscle: fiber-specific adaptation to endurance training // Am. J. Physiol. 1999; 277: R856-R862.
100. Crow J.P., Calingasan N.Y., Chen J., Hill J.L., Beal M.F. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice // Ann. Neurol. 2005; 58: 258-265.
101. Liu R., Li B., Flanagan S.W., Oberley L.W., Gozal D., Qiu M. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival // J. Neurochem. 2002; 80: 488-500.
102. Orrell R.W., Lane R.J., Ross M. Antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease // Cochrane Database Syst. Rev. 2005; 1: CD002829.
103. Lino M.M., Schneider C., Caroni P. Accumulation of SOD1 mutants in postnatal moto-neurons does not cause motoneuron pathology or motoneuron disease // J. Neurosci. 2002; 22: 4825-4832.
104. Pramatarova A., Laganiere J., Roussel J., Brisebois K., Rouleau G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment // J. Neurosci. 2001; 21: 3369-3374.
105. Boillee S., Yamanaka K., Lobsiger C.S., Copeland N.G. et al. Onset and progression in inherited ALS determined by motor neurons and microglia // Science 2006; 312: 1389-1392.
106. Clement A., Nguyen M., Roberts E., Garcia M. et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice // Science 2003; 302: 113-117.
107. Hall E.D., Oostveen J.A., Gurney M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS // Glia 1998; 23: 249-256.
108. Howland D.S., Liu J., She Y. et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) // Proc. Natl. Acad. Sci. USA 2002; 99: 1604-1609.
109. Di Giorgio F.P., Carrasco M.A., Siao M.C., Maniatis T., Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model // Nat. Neurosci. 2007; 10: 608-614.
110. Nagai M., Re D.B., Nagata T., Chalazonitis A. et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons // Nat. Neurosci. 2007; 10: 615-622.
111. De Winter F., Vo T., Stam F.J., Wisman L.A. et al. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease // Mol. Cell. Neurosci. 2006; 32: 102-117.
112. Turner B.J., Lopes E.C., Cheema S.S. Neuromuscular accumulation of mutant superoxide dismutase 1 aggregates in a transgenic mouse model of familial amyotrophic lateral sclerosis // Neurosci. Lett. 2003; 350: 132-136.
113. Taylor A.R., Gifondorwa D.J., Newbern J.M, Robin-son M.B. et al. Astrocyte and muscle-derived secreted factors differentially regulate motoneuron survival // J. Neurosci. 2007; 27: 634-644.
114. Dupuis L., Gonzalez de Aguilar J.L., di Scala F., Rene F., de Tapia M. et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis // Neurobiol. Dis. 2002; 10: 358-365.
115. Jokic N., Gonzalez de Aguilar J.L., Dimou L. et al. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model // EMBO Rep. 2006; 7: 1162-1167.
116. Miller T.M., Kim S.H., Yamanaka K. et al. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis // Proc. Natl. Acad. Sci. USA 2006; 103: 19546-19551.
117. Holzbaur E.L., Howland D.S., Weber N. et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis // Neurobiol. Dis. 2006; 23: 697-707.
118. Dobrowolny G., Giacinti C., Pelosi L., Nicoletti C. et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model // J. Cell. Biol. 2005; 168: 193-199.
119. Cifuentes-Diaz C., Frugier T., Tiziano F.D., Lacene E. et al. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy // J. Cell. Biol. 2001; 152: 1107-1114.
120. Rosen D., Siddique T., Patterson D., Figlewicz D., Sapp P. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis // Nature 1993; 362: 59-62.
121. Ranta S., Zhang Y., Ross B., Lonka L., Takkunen E. et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8 // Nat. Genet. 1999; 23: 233-236.
122. Lefebvre S., Burglen L., Reboullet S., Clermont O. et al. Identification and characterization of a spinal muscular atrophy-determining gene // Cell 1995; 80: 155-165.
123. McWhorter M.L., Monani U.R., Burghes A.H., Beat-tie C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding // J. Cell. Biol. 2003; 162: 919-931.
124. Rich M.M., Wang X., Cope T.C., Pinter M.J. Reduced neuromuscular quantal content with normal synaptic release time course and depression in canine motor neuron disease // J. Neurophysiol. 2002; 88: 3305-3314.
125. La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy // Nature 1991; 352: 77-79.
126. Katsuno M., Adachi H., Minamiyama M., Waza M. et al. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration // J. Neurosci. 2006; 26: 12106-12117.
127. Chambers D.M., Peters J., Abbott C.M. The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor lalpha, encoded by the Eefla2 gene // Proc. Natl. Acad. Sci. USA 1998; 95: 4463-4468.
128. Newbery H.J., Gillingwater T.H., Dharmasaroja P. et al. Progressive loss of motor neuron function in wasted mice: effects of a spontaneous null mutation in the gene for the eEF1 A2 translation factor // J. Neuropathol. Exp. Neurol. 2005; 64: 295-303.
129. Schmitt-John T., Drepper C., Mussmann A., Hahn P. et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse // Nat. Genet. 2005; 37: 1213-1215.
130. Blondet B., Carpentier G., Ait-Ikhlef A., Murawsky M., Rieger F. Motoneuron morphological alterations before and after the onset of the disease in the wobbler mouse // Brain Res. 2002; 930: 53-57.
131. Oosthuyse B., Moons L., Storkebaum E., Beck H. et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration // Nat. Genet. 2001; 28: 131-138.
132. Miura H., Oda K., Endo C., Yamazaki K., Shibasaki H., Kikuchi T. Progressive degeneration of motor nerve terminals in GAD mutant mouse with hereditary sensory axonopathy // Neuropathol. Appl. Neurobiol. 1993; 19: 41-51.
133. Liedtke W., Leman E.E., Fyffe R.E., Raine C.S., Schubart U.K. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems // Am. J. Pathol. 2002; 160: 469-480.
134. Dewil M., dela Cruz V.F., Van Den Bosch L., Robberecht W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1G93A -induced motor neuron death // Neurobiol. Dis. 2007; 26: 332-341.
135. Rouaux C., Panteleeva I., Rene F., Gonzalez de Aguilar J.L. et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model // J. Neurosci. 2007; 27: 5535-5545.
Полинейропатия — это патология периферической нервной системы, которая развивается в результате диффузного повреждения периферических нервов и их аксонов. Отсюда и название болезни. В ее основе — генерализованное поражение осевого цилиндра периферических нервов.

Что такое аксональная полинейропатия
Полиневропатия (второе название — полиневрит) — это клинический синдром, который возникает из-за ряда факторов, влияющих на периферическую нервную систему, и отличается размытыми патогенетическими изменениями. Заболевание занимает одно из лидирующих мест в перечне недугов периферической нервной системы, уступая первенство только вертеброгенной патологии, превосходящей по сложности клинической картины и последствиям, развивающимся из-за нее.
Аскональная полинейропатия считается междисциплинарной проблемой, с ней часто сталкиваются доктора различных специализаций. В первую очередь с данным заболеванием обращаются к неврологу. Частота возникающего синдрома неизвестна, так как отсутствуют статистические данные.
На данный момент известны всего три важных патоморфологических механизма, которые лежат в истоках формирования полинейропатии:
- валлеровская дегенерация;
- первичная демиелинизация;
- первичная аксонопатия.
В соответствии с иммунологической теорией полинейропатия является результатом перекрестного образования иммунных глобулинов, уничтожающих собственные клетки, в результате чего возникает некроз тканей и мышечное воспаление.
Исследователи выдвигают ряд гипотез возникновения и проблем течения аксональной полинейропатии:
- Сосудистая. Базируется на вовлечении в процесс сосудов, по которым кислород и питательные вещества поступают в периферические нервы. Изменяются характеристики крови по качественному и количественному составу, что может привести к ишемии нервных окончаний.
- Теория оксидативного стресса. Позиционирует формирование болезни со стороны нарушения обмена оксида азота, вследствие чего изменяются калий-натриевые механизмы, лежащие в основе формирования нервного возбуждения и проведения импульсов по нервам.
- Теория деактивации факторов роста нерва. Говорит о том, что болезнь возникает из-за недостатка аксонального транспорта с последующим развитием аксонопатии.
- Иммунологическая. Объясняет развитие заболевания в результате перекрестного образования антител к структурам периферической нервной системы, которое сопровождается аутоиммунным воспалением, а затем и некрозом нервов.
Даже при использовании ультрасовременных методов диагностики сложно найти достоверную причину патологии, выяснить ее получается только у 50-70% пострадавших.
Факторов возникновения полинейропатии нижних конечностей по аксональному типу очень много. Однако даже инновационные способы исследования не позволяют установить истинную этиологию заболевания.
Мнение эксперта
Автор: Алексей Владимирович Васильев
Руководитель НПЦ болезни двигательного нейрона/БАС, кандидат медицинских наук, врач высшей категории
Аксональная полинейропатия — это одно из самых опасных неврологических заболеваний, сопровождающееся поражением периферической нервной системы. При болезни разрушаются периферические нервные волокна.
Причин возникновение аксональной полинейропатии несколько. Самые распространенные:
- Сахарный диабет нарушает структуру крови, питающей нервы, в свою очередь происходит сбой в обменных процессах.
- Длительный дефицит витаминов В. Именно они максимально важны для правильной работы нервной системы, поэтому долгая нехватка способна привести к аксональной полинейропатии.
- Воздействие токсинов на организм. К ним относят разнообразные отравляющие вещества, например, алкоголь, а также ВИЧ. При отравлении опасными веществами заболевание может развиться уже через несколько дней.
- Наследственный фактор.
- Синдром Гийена-Барре.
- Различные травмы, к которым также относится длительное сдавливание нервов, которое характерно при грыже или остеохондрозе.
Лечение аксональной полинейропатии обязательно должно быть комплексным, иначе нужного эффекта достичь не удастся. Категорически запрещается заниматься самолечением и при возникновении первых же симптомов нужно срочно обратиться к доктору. Врачи Юсуповской больницы подбирают лечение индивидуально для каждого пациента. В зависимости от тяжести патологии и симптоматики назначается комплексное лечение под наблюдением опытных специалистов.

Причины
Самые распространенные причины возникновения аксональной полинейропатии нижних конечностей:
- истощение организма;
- длительный недостаток витаминов группы В;
- недуги, ведущие к дистрофии;
- острые инфекции;
- токсическое поражение ртутью, свинцом, кадмием, угарным газом, спиртными напитками, метиловым спиртом, фосфорорганическими соединениями, медицинскими препаратами, принимаемыми без согласования с врачом;
- болезни сердечно-сосудистой, кроветворной, кровеносной и лимфатической систем;
- эндокринологические патологии, в том числе инсулинозависимость.
Главными факторами, которые провоцируют развитие моторной или сенсомоторной аксональной полинейропатии, являются:
- эндогенная интоксикация при почечной недостаточности;
- аутоиммунные процессы, протекающие в организме;
- амилоидоз;
- вдыхание токсических веществ или паров.
Также болезнь может быть обусловлена наследственностью.
Нехватка в организме витаминов группы В, а в особенности пиридоксина и цианокобаламина, крайне негативно воздействует на проводимость нервных и моторных волокон и может вызывать сенсорную аксональную полинейропатию нижних конечностей. Это же происходит при хронической алкогольной интоксикации, глистной инвазии, заболеваниях желудочно-кишечного тракта, которые ухудшают скорость всасывания.
Токсическое отравление лекарственными препаратами, аминогликозидами, золотыми солями и висмутом занимают большой процент в структуре факторов аксональной невропатии.
У пациентов с сахарным диабетом нарушена функция периферических нервов из-за нейротоксичности кетоновых тел, то есть метаболитов жирных кислот. Происходит это из-за невозможности организма использовать глюкозу как главный источник энергии. Поэтому вместо нее окисляются жиры.
При аутоиммунных заболеваниях, протекающих в организме, иммунная система человека атакует собственные нервные волокна, воспринимая их как источник опасности. Это происходит из-за провокации иммунитета, возникающей при неосторожном приеме иммуностимулирующих медикаментов и нетрадиционных методик лечения. Поэтому у людей, которые склонны к возникновению аутоиммунных заболеваний, пусковыми факторами аксональной полинейропатии являются:
- иммуностимуляторы;
- вакцины;
- аутогемотерапия.
При амилоидозе в организме накапливается такой белок, как амилоид. Именно он нарушает основные функции нервных волокон.
Первые признаки
Заболевание обычно начинает развиваться с поражения толстых или тонких нервных волокон. Зачастую аксональная полинейропатия имеет дистальное симметричное распределение на кисти или стопы. Нейропатия чаще всего сначала поражает нижние конечности, а затем симметрично распространяется вверх по телу. К самым частым первичным симптомам поражения относят:
- мышечную слабость;
- болевой синдром в конечностях;
- жжение;
- ощущение ползания мурашек;
- онемение кожных покровов.
Симптоматика ярче всего проявляется в вечернее и ночное время суток.
Симптомы
Врачи подразделяют хроническое, острое и подострое течение аксональной полинейропатии. Заболевание подразделяется на два вида: первично-аксональный и демиелинизирующий. В ходе течения болезни к ней присовокупляется демиелинизация, а затем и вторично аксональный компонент.
К основным проявлениям недуга относятся:
- вялость в мышцах ног или рук;
- спастический паралич конечностей;
- чувство подергивания в мышечных волокнах;
- головокружение при резкой перемене положения тела;
- отек конечностей;
- жжение;
- покалывание;
- ощущение ползания мурашек;
- снижение чувствительности кожных покровов к высокой или низкой температуре, боли и касаниям;
- нарушение ясности речи;
- проблемы с координацией.
Вегетативными признаками сенсомоторной полинейропатии асконального типа считаются следующие симптомы:
- учащенный или, напротив, замедленный сердечный ритм;
- неумеренное потоотделение;
- чрезмерная сухость кожи;
- изменение цвета кожных покровов;
- нарушение эякуляции;
- эректильная дисфункция;
- проблемы с мочеиспусканием;
- сбой двигательных функций желудочно-кишечного тракта;
- повышенное слюнотечение или, наоборот, сухость во рту;
- расстройство аккомодации глаза.
Заболевание проявляется в нарушениях функций поврежденных нервов. Именно периферические нервные волокна отвечают за двигательные функции мышечной ткани, чувствительность, а также оказывают вегетативное воздействие, то есть регулируют сосудистый тонус.
Для нарушения функции проводимости нервов характерны расстройства чувствительности, например:
- чувство ползания мурашек;
- гиперестезия, то есть увеличение чувствительности кожи к внешним раздражителям;
- гипестезия, то есть уменьшение чувствительности;
- отсутствие ощущения собственных конечностей.
Когда поражены вегетативные волокна, то из-под контроля выходит регуляция сосудистого тонуса. При аксонально-демиелинизирующей полинейропатии наступает сдавление капилляров, из-за чего ткани отекают. Нижние, а затем и верхние конечности из-за скапливания в них жидкости существенно увеличиваются в размерах. Так как при полинейропатии нижних конечностей основное количество крови накапливается именно в пораженных областях тела, то у пациента возникает стойкое головокружение при принятии вертикального положения. Из-за того, что пропадает трофическая функция, могут возникнуть эрозивно-язвенные поражения нижних конечностей.
Аксональная моторная полинейропатия проявляется в двигательных нарушениях верхних и нижних конечностей. Когда моторные волокна, отвечающие за движения рук и ног, повреждены, то наступает полный или частичный паралич мышц. Обездвиживание может проявляться совершенно нетипично — может ощущаться как скованность мышечных волокон, так и чрезмерная их расслабленность. При средней степени поражения ослаблен мышечный тонус.
В ходе течения заболевания могут быть усилены или ослаблены сухожильные и надкостничные рефлексы. В редких случаях доктор-невролог их не наблюдает. При болезни часто могут быть поражены черепные нервы, которые проявляются следующими нарушениями:
- глухотой;
- онемением подъязычных мышц и мускулатуры языка;
- невозможностью проглотить еду или жидкость из-за проблем с глотательным рефлексом.
Когда поражен тройничный, лицевой или глазодвигательный нерв, изменяется чувствительность кожных покровов, развиваются параличи, возникает асимметрия лица и подергивание мышц. Иногда при диагностированной аксонально-демиелинизирующей полинейропатии поражения верхних или нижних конечностей могут быть асимметричными. Такое случается при множественной мононейропатии, когда коленные, ахилловы и карпорадиальные рефлексы несимметричны.
Диагностика
Главной методикой исследования, которая позволяет обнаружить локализацию патологического процесса и степень пораженности нервов, является электронейромиография.
Чтобы определить причину заболевания, врачи назначают следующие анализы:
- определение уровня сахара в плазме крови;
- токсикологические тесты;
- полный анализ мочи и крови;
- выявление уровня холестерина в организме.
Нарушение нервных функций устанавливается при помощи определения температурной, вибрационной и тактильной чувствительности.
При первичном осмотре применяется зрительная методика исследования. То есть врач, к которому обратился с жалобами пострадавший, осматривает и анализирует такие внешние симптомы, как:
- уровень давления крови в верхних и нижних конечностях;
- чувствительность кожных покровов к прикосновениям и температуре;
- наличие всех необходимых рефлексов;
- диагностика отечности;
- изучение внешнего состояния кожи.
Выявить аксональную полинейропатию можно при помощи следующих инструментальных исследований:
- магнитная резонансная томография;
- биопсия нервных волокон;
- электронейромиография.
Лечение аксональной полинейропатии
Лечение аксональной полинейропатии должно быть комплексным и направленным на причину развития заболевания, его механизмы и симптоматику. Гарантией эффективной терапии является своевременное выявление болезни и лечение, которое сопровождается абсолютным отказом от сигарет, алкоголя и наркотических веществ, ведением здорового образа жизни и соблюдением всех рекомендаций врача. В первую очередь проводятся следующие терапевтические мероприятия:
- избавление от токсического воздействия на организм, если оно присутствует;
- антиоксидантная терапия;
- прием препаратов, которые воздействуют на тонус кровеносных сосудов;
- восполнение дефицита витаминов;
- регулярный контроль концентрации глюкозы в плазме крови.
Отдельное внимание уделяется лечению, направленному на купирование острого болевого синдрома.
Если присутствуют периферические парезы, то есть существенное снижение мышечной силы с многократным уменьшением амплитуды движений, то в обязательном порядке показана лечебная физкультура и специальные физические упражнения, направленные на возвращение тонуса мышечным тканям и предотвращение образования различных контрактур. Особенно важна регулярная психологическая поддержка, которая не дает пациенту впасть в депрессию, сопровождающуюся расстройством сна и чрезмерной нервной возбудимостью.
Лечение аксональной полинейропатии — это продолжительный процесс, так как нервные волокна восстанавливаются долго. Поэтому не стоит ожидать моментального выздоровления и возвращения к привычному образу жизни. Медикаментозная терапия включает такие препараты, как:
- обезболивающее;
- глюкокортикоиды;
- витамины группы В;
- антиоксиданты;
- сосудорасширяющие;
- средства, ускоряющие метаболизм и улучшающие микроциркуляцию крови.
Терапия лекарственными препаратами направлена на восстановление функций нервов, улучшение проводимости нервных волокон и скорости передачи сигналов центральной нервной системе.
Лечение следует проводить длительными курсами, которые не стоит прерывать, хоть и эффект от них проявляется не сразу. Чтобы устранить болевые ощущения и расстройство сна, назначают следующие медикаменты:
- антидепрессанты;
- противосудорожные;
- препараты, купирующие аритмию;
- обезболивающие.
Для избавления от боли используют нестероидные противовоспалительные препараты. Но стоит помнить, что применять их можно только короткий промежуток времени, так как длительное употребление может привести к повреждению слизистой оболочки желудочно-кишечного тракта.
К физиотерапевтическим методам лечения аксональной полинейропатии относятся:
- терапия магнитными волнами;
- грязелечение;
- электростимуляция;
- иглоукалывание;
- лечебный массаж;
- физкультура;
- ультрафонофорез;
- гальванотерапия.
Именно лечебная физкультура позволяет сохранить работоспособность мышечных тканей и поддерживать конечности в нужном положении. Регулярные занятия спортом вернут мышцам тонус, гибкость и увеличат амплитуду движений до нормальной.
Прогноз
Если заболевание обнаружено на ранней стадии и комплексно лечится квалифицированными специалистами, то прогноз для жизни и здоровья пациента более чем благоприятный. Стоит вести правильный образ жизни, рацион должен быть богат витаминами и минералами, необходимыми для правильного функционирования организма.
Если долгое время игнорировать болезнь и не предпринимать никаких действий, результат будет плачевным вплоть до полного паралича.
Профилактика
Пациент в обязательном порядке должен совершать профилактические мероприятия, которые помогут избежать рецидива или возникновения опасного заболевания. Они включают в себя обогащение рациона витаминами, регулярный контроль уровня сахара в крови, полный отказ от табакокурения, наркотических веществ и алкогольных напитков.
В целях профилактики болезни рекомендуется:
- носить удобную обувь, которая не пережимает стопу, ухудшая кровоток;
- регулярно осматривать обувь, чтобы избежать образования грибка;
- исключить пешие прогулки на длительные расстояния;
- не стоять долгое время на одном месте;
- мыть ноги прохладной водой или делать контрастные ванночки, что помогает улучшить циркуляцию крови в организме.
Пострадавшим в стадии ремиссии категорически запрещается принимать лекарственные препараты без согласования с лечащим врачом. Важно своевременно лечить воспалительные заболевания, соблюдать меры предосторожности при работе с токсическими веществами, которые оказывают пагубное воздействие на организм, регулярно выполнять лечебные физические упражнения.
From Wikipedia, the free encyclopedia
| Nerve injury | |
|---|---|
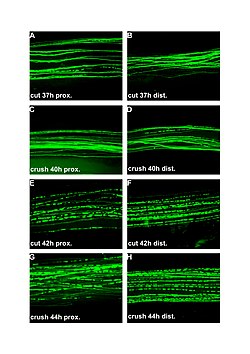 |
|
| Fluorescent micrographs (100x) of Wallerian degeneration in cut and crushed peripheral nerves. Left column is proximal to the injury, right is distal. A and B: 37 hours post cut. C and D: 40 hours post crush. E and F: 42 hours post cut. G and H: 44 hours post crush. | |
| Specialty | Neurology |
Wallerian degeneration is an active process of degeneration that results when a nerve fiber is cut or crushed and the part of the axon distal to the injury (which in most cases is farther from the neuron’s cell body) degenerates.[1] A related process of dying back or retrograde degeneration known as ‘Wallerian-like degeneration’ occurs in many neurodegenerative diseases, especially those where axonal transport is impaired such as ALS and Alzheimer’s disease.[2] Primary culture studies suggest that a failure to deliver sufficient quantities of the essential axonal protein NMNAT2 is a key initiating event.[3][4]
Wallerian degeneration occurs after axonal injury in both the peripheral nervous system (PNS) and central nervous system (CNS). It occurs in the section of the axon distal to the site of injury and usually begins within 24–36 hours of a lesion. Prior to degeneration, the distal section of the axon tends to remain electrically excitable. After injury, the axonal skeleton disintegrates, and the axonal membrane breaks apart. Axonal degeneration is followed by degradation of the myelin sheath and infiltration by macrophages. The macrophages, accompanied by Schwann cells, serve to clear the debris from the degeneration.[5][6]
Schwann cells respond to loss of axons by extrusion of their myelin sheaths, downregulation of myelin genes, dedifferentiation and proliferation. They finally align in tubes (Büngner bands) and express surface molecules that guide regenerating fibers.[7] Within 4 days of the injury, the distal end of the portion of the nerve fiber proximal to the lesion sends out sprouts towards those tubes and these sprouts are attracted by growth factors produced by Schwann cells in the tubes. If a sprout reaches the tube, it grows into it and advances about 1 mm per day, eventually reaching and reinnervating the target tissue. If the sprouts cannot reach the tube, for instance because the gap is too wide or scar tissue has formed, surgery can help to guide the sprouts into the tubes. Regeneration is efficient in the PNS, with near complete recovery in case of lesions that occur close to the distal nerve terminal. However recovery is hardly observed at all in the spinal cord. One crucial difference is that in the CNS, including the spinal cord, myelin sheaths are produced by oligodendrocytes and not by Schwann cells.
History[edit]
Wallerian degeneration is named after Augustus Volney Waller. Waller experimented on frogs in 1850, by severing their glossopharyngeal and hypoglossal nerves. He then observed the distal nerves from the site of injury, which were separated from their cell bodies in the brain stem.[5] Waller described the disintegration of myelin, which he referred to as «medulla», into separate particles of various sizes. The degenerating axons formed droplets that could be stained, thus allowing for studies of the course of individual nerve fibres.
Axonal degeneration[edit]
Although most injury responses include a calcium influx signaling to promote resealing of severed parts, axonal injuries initially lead to acute axonal degeneration (AAD), which is rapid separation of the proximal (the part nearer the cell body) and distal ends within 30 minutes of injury.[8] After separation, dystrophic bulb structures form at both terminals and the transected membranes are sealed.[9] A brief latency phase occurs in the distal segment during which it remains electrically excitable and structurally intact.[10] Degeneration follows with swelling of the axolemma, and eventually the formation of bead-like axonal spheroids. The process takes roughly 24 hours in the PNS, and longer in the CNS. The signaling pathways leading to axolemma degeneration are currently poorly understood. However, research has shown that this AAD process is calcium–independent.[11]
Granular disintegration of the axonal cytoskeleton and inner organelles occurs after axolemma degradation. Early changes include accumulation of mitochondria in the paranodal regions at the site of injury. Endoplasmic reticulum degrades and mitochondria swell up and eventually disintegrate. The depolymerization of microtubules occurs and is soon followed by degradation of the neurofilaments and other cytoskeleton components. The disintegration is dependent on Ubiquitin and Calpain proteases (caused by influx of calcium ion), suggesting that axonal degeneration is an active process and not a passive one as previously misunderstood.[12] Thus the axon undergoes complete fragmentation. The rate of degradation is dependent on the type of injury and is also slower in the CNS than in the PNS. Another factor that affects degradation rate is the diameter of the axon: larger axons require a longer time for the cytoskeleton to degrade and thus take a longer time to degenerate.
Myelin clearance[edit]
Myelin is a phospholipid membrane that wraps around axons to provide them with insulation. It is produced by Schwann cells in the PNS, and by oligodendrocytes in the CNS. Myelin clearance is the next step in Wallerian degeneration following axonal degeneration. The cleaning up of myelin debris is different for PNS and CNS.
PNS is much faster and efficient at clearing myelin debris in comparison to CNS, and Schwann cells are the primary cause of this difference. Another key aspect is the change in permeability of the blood-tissue barrier in the two systems. In PNS, the permeability increases throughout the distal stump, but the barrier disruption in CNS is limited to just the site of injury.[11]
Clearance in PNS[edit]
The response of Schwann cells to axonal injury is rapid. The time period of response is estimated to be prior to the onset of axonal degeneration. Neuregulins are believed to be responsible for the rapid activation. They activate ErbB2 receptors in the Schwann cell microvilli, which results in the activation of the mitogen-activated protein kinase (MAPK).[13] Although MAPK activity is observed, the injury sensing mechanism of Schwann cells is
yet to be fully understood. The ‘sensing’ is followed by decreased synthesis of myelin lipids and eventually stops within 48 hrs. The myelin sheaths separate from the axons at the Schmidt-Lanterman incisures first and then rapidly deteriorate and shorten to form bead-like structures. Schwann cells continue to clear up the myelin debris by degrading their own myelin, phagocytose extracellular myelin and attract macrophages to myelin debris for further phagocytosis.[11] However, the macrophages are not attracted to the region for the first few days; hence the Schwann cells take the major role in myelin cleaning until then.
Schwann cells have been observed to recruit macrophages by release of cytokines and chemokines after sensing of axonal injury. The recruitment of macrophages helps improve the clearing rate of myelin debris. The resident macrophages present in the nerves release further chemokines and cytokines to attract further macrophages.
The degenerating nerve also produce macrophage chemotactic molecules. Another source of macrophage recruitment factors is serum. Delayed macrophage recruitment was observed in B-cell deficient mice lacking serum antibodies.[11] These signaling molecules together cause an influx of macrophages, which peaks during the third week after injury. While Schwann cells mediate the initial stage of myelin debris clean up, macrophages come in to finish the job. Macrophages are facilitated by opsonins, which label debris for removal. The 3 major groups found in serum include complement, pentraxins, and antibodies. However, only complement has shown to help in myelin debris phagocytosis.[14]
Murinson et al. (2005)[15] observed that non-myelinated or myelinated Schwann cells in contact with an injured
axon enter cell cycle thus leading to proliferation. Observed time duration for
Schwann cell divisions were approximately 3 days after injury.[16]
Possible sources of proliferation signal are attributed to the ErbB2 receptors and the ErbB3 receptors. This proliferation could further enhance the myelin cleaning rates and plays an essential role in regeneration of axons observed in PNS. Schwann cells emit growth factors that attract new axonal sprouts growing from the proximal stump after complete degeneration of the injured distal stump. This leads to possible reinnervation of the target cell or organ. However, the reinnervation is not necessarily perfect, as possible misleading occurs during reinnervation of the proximal axons to target cells.
Clearance in CNS[edit]
In comparison to Schwann cells, oligodendrocytes require axon signals to survive. In their developmental stages, oligodendrocytes that fail to make contact to axon and receive axon signals undergo apoptosis.[17]
Experiments in Wallerian degeneration have shown that upon injury oligodendrocytes either undergo programmed cell death or enter a state of rest. Therefore, unlike Schwann cells, oligodendrocytes fail to clean up the myelin sheaths and their debris. In experiments conducted on rats,[18] myelin sheaths were found for up to 22 months. Therefore, CNS rates of myelin sheath clearance are very slow and could possibly be the cause for hindrance in the regeneration capabilities of the CNS axons as no growth factors are available to attract the proximal axons. Another feature that results eventually is Glial scar formation. This further hinders chances for regeneration and reinnervation.
Oligodendrocytes fail to recruit macrophages for debris removal. Macrophage entry in general into CNS site of injury is very slow. In contrast to PNS, Microglia play a vital role in CNS wallerian degeneration. However, their recruitment is slower in comparison to macrophage recruitment in PNS by approximately 3 days. Further, microglia might be activated but hypertrophy, and fail to transform into fully phagocytic cells. Those microglia that do transform, clear out the debris effectively. Differentiating phagocytic microglia can be accomplished by testing for expression of Major histocompatibility complex (MHC) class I and II during wallerian degeneration.[19] The rate of clearance is very slow among microglia in comparison to macrophages. Possible source for variations in clearance rates could include lack of opsonin activity around microglia, and the lack of increased permeability in the blood–brain barrier. The decreased permeability could further hinder macrophage infiltration to the site of injury.[11]
These findings have suggested that the delay in Wallerian degeneration in CNS in comparison to PNS is caused not due to a delay in axonal degeneration, but rather is due to the difference in clearance rates of myelin in CNS and PNS.[20]
Regeneration[edit]
Regeneration follows degeneration. Regeneration is rapid in PNS, allowing for rates of up to 1 millimeter a day of regrowth.[21] Grafts may also be needed to allow for appropriate reinnervation. It is supported by Schwann cells through growth factors release. CNS regeneration is much slower, and is almost absent in most vertebrate species. The primary cause for this could be the delay in clearing up myelin debris. Myelin debris, present in CNS or PNS, contains several inhibitory factors. The prolonged presence of myelin debris in CNS could possibly hinder the regeneration.[22] An experiment conducted on newts, animals that have fast CNS axon regeneration capabilities, found that Wallerian degeneration of an optic nerve injury took up to 10 to 14 days on average, further suggesting that slow clearance inhibits regeneration.[23]
Schwann cells and endoneural fibroblasts in PNS[edit]
In healthy nerves, nerve growth factor (NGF) is produced in very small amounts. However, upon injury, NGF mRNA expression increases by five to seven-fold within a period of 14 days. Nerve fibroblasts and Schwann cells play an important role in increased expression of NGF mRNA.[24] Macrophages also stimulate Schwann cells and fibroblasts to produce NGF via macrophage-derived interleukin-1.[25] Other neurotrophic molecules produced by Schwann cells and fibroblasts together include brain-derived neurotrophic factor, glial cell line-derived neurotrophic factor, ciliary neurotrophic factor, leukemia inhibitory factor, insulin-like growth factor, and fibroblast growth factor. These factors together create a favorable environment for axonal growth and regeneration.[11] Apart from growth factors, Schwann cells also provide structural guidance to further enhance regeneration. During their proliferation phase, Schwann cells begin to form a line of cells called Bands of Bungner within the basal laminar tube. Axons have been observed to regenerate in close association to these cells.[26] Schwann cells upregulate the production of cell surface adhesion molecule ninjurin further promoting growth.[27] These lines of cell guide the axon regeneration in proper direction. The possible source of error that could result from this is possible mismatching of the target cells as discussed earlier.
Due to lack of such favorable promoting factors in CNS, regeneration is stunted in CNS.
Wallerian degeneration slow[edit]
Mice belonging to the strain C57BL/Wlds have delayed Wallerian degeneration,[28] and, thus, allow for the study of the roles of various cell types and the underlying cellular and molecular processes. Current understanding of the process has been possible via experimentation on the Wlds strain of mice. The mutation occurred first in mice in Harlan-Olac, a laboratory producing animals the United Kingdom. The Wlds mutation is an autosomal-dominant mutation occurring in the mouse chromosome 4.[29][30] The gene mutation is an 85-kb tandem triplication, occurring naturally. The mutated region contains two associated genes: nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) and ubiquitination factor e4b (UBE4B). A linker region encoding 18 amino acids is also part of the mutation.[6] The protective effect of the WldS protein has been shown to be due to the NMNAT1 region’s NAD+ synthesizing active site.[31]
Although the protein created localizes within the nucleus and is barely detectable in axons, studies suggest that its protective effect is due to its presence in axonal and terminal compartments.[32][33] The protection provided by the WldS protein is intrinsic to the neurons and not surrounding support cells, and is only locally protective of the axon, indicating an intracellular pathway is responsible for mediating Wallerian degeneration.[34][35]
Effects of the WldS mutation[edit]
The mutation causes no harm to the mouse. The only known effect is that the Wallerian degeneration is delayed by up to three weeks on average after injury of a nerve. At first, it was suspected that the Wlds mutation slows down the macrophage infiltration, but recent studies suggest that the mutation protects axons rather than slowing down the macrophages.[6] The process by which the axonal protection is achieved is poorly understood. However, studies suggest that the Wlds mutation leads to increased NMNAT1 activity, which leads to increased NAD+ synthesis.[31] This in turn activates SIRT1-dependent process within the nucleus, causing changes in gene transcription.[31] NAD+ by itself may provide added axonal protection by increasing the axon’s energy resources.[36] More recent work, however, raises doubt that either NMNAT1 or NAD+ can substitute for the full length Wlds gene.[37] These authors demonstrated by both in vitro and in vivo methods that the protective effect of overexpression of NMNAT1 or the addition of NAD+ did not protect axons from degeneration. However, later studies showed that NMNAT1 is protective when combined with an axonal targeting peptide, suggesting that the key to the protection provided by WldS was the combination of NMNAT1’s activity and the axonal localization provided by the N-terminal domain of the chimeric protein.[38]
The provided axonal protection delays the onset of Wallerian degeneration. Schwann cell activation should therefore be delayed, as they would not detect axonal degradation signals from ErbB2 receptors. In experiments on Wlds mutated mice, macrophage infiltration was considerably delayed by up to six to eight days.[39] However, once the axonal degradation has begun, degeneration takes its normal course, and, respective of the nervous system, degradation follows at the above-described rates. Possible effects of this late onset are weaker regenerative abilities in the mice. Studies indicate that regeneration may be impaired in WldS mice, but this is likely a result of the environment being unfavorable for regeneration due to the continued existence of the undegenerated distal fiber, whereas normally debris is cleared, making way for new growth.[40]
SARM1[edit]
The Wallerian degeneration pathway has been further illuminated by the discovery that sterile alpha and TIR motif containing 1 (SARM1) protein plays a central role in the Wallerian degeneration pathway. The gene was first identified in a Drosophila melanogaster mutagenesis screen, and subsequently knockouts of its homologue in mice showed robust protection of transected axons comparable to that of WldS.[41][42]
SARM1 catalyzes the synthesis and hydrolysis of cyclic ADP-ribose (cADPR) from NAD+ to ADP-ribose.[43] SARM1 activation locally triggers a rapid collapse of NAD+ levels in the distal section of the injured axon, which then undergoes degeneration.[44] This collapse in NAD+ levels was later shown to be due to SARM1’s TIR domain having intrinsic NAD+ cleavage activity.[45] The SARM1 protein has four domains, a mitochondrial localization signal, an auto-inhibitory N-terminus region consisting of armadillo/HEAT motifs, two sterile alpha motifs responsible for multimerization, and a C-terminus Toll/Interleukin-1 receptor that possesses enzymatic activity.[45] Activation of SARM1 is sufficient to collapse NAD+ levels and initiate the Wallerian degeneration pathway.[44]
The activity of SARM1 helps to explain the protective nature of the survival factor NMNAT2, as NMNAT enzymes have been shown to prevent SARM1-mediated depletion of NAD+.[46] This relationship is further supported by the fact that mice lacking NMNAT2, which are normally not viable, are completely rescued by SARM1 deletion, placing NMNAT2 activity upstream of SARM1.[47] Other pro-degeneration signaling pathways, such as the MAP kinase pathway, have been linked to SARM1 activation. MAPK signaling has been shown to promote the loss of NMNAT2, thereby promoting SARM1 activation, although SARM1 activation also triggers the MAP kinase cascade, indicating some form of feedback loop exists.[48][49] One explanation for the protective effect of the WldS mutation is that the NMNAT1 region, which is normally localized to the soma, substitutes for the labile survival factor NMNAT2 to prevent SARM1 activation when the N-terminal Ube4 region of the WldS protein localizes it to the axon. The fact that the enhanced survival of WldS axons is due to the slower turnover of WldS compared to NMNAT2 also helps explain why SARM1 knockout confers longer protection, as SARM1 will be completely inactive regardless of inhibitor activity whereas WldS will eventually be degraded. Possibles implications of the SARM1 pathway in regard to human health may be found in animal models which exhibit traumatic brain injury, as mice which contain Sarm1 deletions in addition to WldS show decreased axonal damage following injury.[50] Specific mutations in NMNAT2 have linked the Wallerian degeneration mechanism to two neurological diseases.
See also[edit]
- Axonotmesis
- Connective tissue in the peripheral nervous system
- Diffuse axonal injury
- Digestion chambers
- Nerve injury
- Neuroregeneration
- Peripheral nerve injury
- Primary and secondary brain injury
- Seddon’s classification
- Spinal cord injury research
References[edit]
- ^ Trauma and Wallerian Degeneration, University of California, San Francisco
- ^ Coleman MP, Freeman MR (1 June 2010). «Wallerian degeneration, wld(s), and nmnat». Annual Review of Neuroscience. 33 (1): 245–67. doi:10.1146/annurev-neuro-060909-153248. PMC 5223592. PMID 20345246.
- ^ Gilley J, Coleman MP (January 2010). «Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons». PLOS Biology. 8 (1): e1000300. doi:10.1371/journal.pbio.1000300. PMC 2811159. PMID 20126265.
- ^ Brazill JM, Li C, Zhu Y, Zhai RG (2017). «NMNAT: It’s an NAD + Synthase… It’s a Chaperone… It’s a Neuroprotector». Current Opinion in Genetics & Development. 44: 156–162. doi:10.1016/j.gde.2017.03.014. PMC 5515290. PMID 28445802.
- ^ a b Waller A (1 January 1850). «Experiments on the Section of the Glossopharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres». Philosophical Transactions of the Royal Society of London. 140: 423–429. doi:10.1098/rstl.1850.0021. JSTOR 108444.
- ^ a b c Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH (August 1998). «An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse». Proceedings of the National Academy of Sciences of the United States of America. 95 (17): 9985–90. Bibcode:1998PNAS…95.9985C. doi:10.1073/pnas.95.17.9985. PMC 21448. PMID 9707587.
- ^ Stoll G, Müller HW (April 1999). «Nerve injury, axonal degeneration and neural regeneration: basic insights». Brain Pathology. 9 (2): 313–25. doi:10.1111/j.1750-3639.1999.tb00229.x. PMC 8098499. PMID 10219748. S2CID 24140507.
- ^ Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (May 2005). «In vivo imaging of axonal degeneration and regeneration in the injured spinal cord». Nature Medicine. 11 (5): 572–7. doi:10.1038/nm1229. PMID 15821747. S2CID 25287010.
- ^ Eddleman CS, Ballinger ML, Smyers ME, Fishman HM, Bittner GD (June 1998). «Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury». The Journal of Neuroscience. 18 (11): 4029–41. doi:10.1523/JNEUROSCI.18-11-04029.1998. PMC 6792792. PMID 9592084.
- ^ Wang JT, Medress ZA, Barres BA (January 2012). «Axon degeneration: molecular mechanisms of a self-destruction pathway». The Journal of Cell Biology. 196 (1): 7–18. doi:10.1083/jcb.201108111. PMC 3255986. PMID 22232700.
- ^ a b c d e f Vargas ME, Barres BA (1 July 2007). «Why is Wallerian degeneration in the CNS so slow?». Annual Review of Neuroscience. 30 (1): 153–79. doi:10.1146/annurev.neuro.30.051606.094354. PMID 17506644.
- ^ Zimmerman UP, Schlaepfer WW (March 1984). «Multiple forms of Ca-activated protease from rat brain and muscle». The Journal of Biological Chemistry. 259 (5): 3210–8. doi:10.1016/S0021-9258(17)43282-0. PMID 6321500.
- ^ Guertin AD, Zhang DP, Mak KS, Alberta JA, Kim HA (March 2005). «Microanatomy of axon/glial signaling during Wallerian degeneration». The Journal of Neuroscience. 25 (13): 3478–87. doi:10.1523/JNEUROSCI.3766-04.2005. PMC 6724908. PMID 15800203.
- ^ Dailey AT, Vellino AM, Benthem L, Silver J, Kliot M (September 1998). «Complement depletion reduces macrophage infiltration and ctivation during Wallerian degeneration and axonal regeneration». The Journal of Neuroscience. 18 (17): 6713–22. doi:10.1523/JNEUROSCI.18-17-06713.1998. PMC 6792968. PMID 9712643.
- ^ Murinson BB, Archer DR, Li Y, Griffin JW (February 2005). «Degeneration of myelinated efferent fibers prompts mitosis in Remak Schwann cells of uninjured C-fiber afferents». The Journal of Neuroscience. 25 (5): 1179–87. doi:10.1523/JNEUROSCI.1372-04.2005. PMC 6725954. PMID 15689554.
- ^ Liu HM, Yang LH, Yang YJ (July 1995). «Schwann cell properties: 3. C-fos expression, bFGF production, phagocytosis and proliferation during Wallerian degeneration». Journal of Neuropathology and Experimental Neurology. 54 (4): 487–96. doi:10.1097/00005072-199507000-00002. PMID 7602323. S2CID 25055891.
- ^ Barres BA, Jacobson MD, Schmid R, Sendtner M, Raff MC (August 1993). «Does oligodendrocyte survival depend on axons?». Current Biology. 3 (8): 489–97. doi:10.1016/0960-9822(93)90039-Q. PMID 15335686. S2CID 39909326.
- ^ Ludwin SK (31 May 1990). «Oligodendrocyte survival in Wallerian degeneration». Acta Neuropathologica. 80 (2): 184–91. doi:10.1007/BF00308922. PMID 1697140. S2CID 36103242.
- ^ Koshinaga M, Whittemore SR (April 1995). «The temporal and spatial activation of microglia in fiber tracts undergoing anterograde and retrograde degeneration following spinal cord lesion». Journal of Neurotrauma. 12 (2): 209–22. doi:10.1089/neu.1995.12.209. PMID 7629867.
- ^ George R, Griffin JW (October 1994). «Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model». Experimental Neurology. 129 (2): 225–36. doi:10.1006/exnr.1994.1164. PMID 7957737. S2CID 40089749.
- ^ Lundy-Ekman L (2007). Neuroscience: Fundamentals for Rehabilitation (3rd ed.). Saunders. ISBN 978-1-4160-2578-8.
- ^ He Z, Koprivica V (21 July 2004). «The Nogo signaling pathway for regeneration block». Annual Review of Neuroscience. 27 (1): 341–68. doi:10.1146/annurev.neuro.27.070203.144340. PMID 15217336.
- ^ Turner JE, Glaze KA (March 1977). «The early stages of Wallerian degeneration in the severed optic nerve of the newt (Triturus viridescens)». The Anatomical Record. 187 (3): 291–310. doi:10.1002/ar.1091870303. PMID 851236. S2CID 2028827.
- ^ Heumann R, Korsching S, Bandtlow C, Thoenen H (June 1987). «Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection» (PDF). The Journal of Cell Biology. 104 (6): 1623–31. doi:10.1083/jcb.104.6.1623. PMC 2114490. PMID 3034917.
- ^ Lindholm D, Heumann R, Hengerer B, Thoenen H (November 1988). «Interleukin 1 increases stability and transcription of mRNA encoding nerve growth factor in cultured rat fibroblasts». The Journal of Biological Chemistry. 263 (31): 16348–51. doi:10.1016/S0021-9258(18)37599-9. PMID 3263368.
- ^ Thomas PK, King RH (October 1974). «The degeneration of unmyelinated axons following nerve section: an ultrastructural study». Journal of Neurocytology. 3 (4): 497–512. doi:10.1007/BF01098736. PMID 4436692. S2CID 37385200.
- ^ Araki T, Milbrandt J (August 1996). «Ninjurin, a novel adhesion molecule, is induced by nerve injury and promotes axonal growth». Neuron. 17 (2): 353–61. doi:10.1016/S0896-6273(00)80166-X. PMID 8780658. S2CID 12471778.
- ^ Perry VH, Brown MC, Tsao JW (1 October 1992). «The Effectiveness of the Gene Which Slows the Rate of Wallerian Degeneration in C57BL/Ola Mice Declines With Age». The European Journal of Neuroscience. 4 (10): 1000–2. doi:10.1111/j.1460-9568.1992.tb00126.x. PMID 12106435. S2CID 24786532.
- ^ Perry, V. H., Lunn, E. R., Brown, M. C., Cahusac, S. and Gordon, S. (1990), Evidence that the Rate of Wallerian Degeneration is Controlled by a Single Autosomal Dominant Gene. European Journal of Neuroscience, 2: 408-413. https://doi.org/10.1111/j.1460-9568.1990.tb00433.x
- ^ Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH (October 1993). «A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4». Proceedings of the National Academy of Sciences of the United States of America. 90 (20): 9717–20. Bibcode:1993PNAS…90.9717L. doi:10.1073/pnas.90.20.9717. PMC 47641. PMID 8415768.
- ^ a b c Araki T, Sasaki Y, Milbrandt J (August 2004). «Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration». Science. 305 (5686): 1010–3. Bibcode:2004Sci…305.1010A. doi:10.1126/science.1098014. PMID 15310905. S2CID 32370137.
- ^ Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP (December 2001). «Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene». Nature Neuroscience. 4 (12): 1199–206. doi:10.1038/nn770. hdl:1842/737. PMID 11770485. S2CID 8316115.
- ^ Beirowski B, Babetto E, Gilley J, Mazzola F, Conforti L, Janeckova L, Magni G, Ribchester RR, Coleman MP (January 2009). «Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo». The Journal of Neuroscience. 29 (3): 653–68. doi:10.1523/JNEUROSCI.3814-08.2009. PMC 6665162. PMID 19158292.
- ^ Glass JD, Brushart TM, George EB, Griffin JW (May 1993). «Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon». Journal of Neurocytology. 22 (5): 311–21. doi:10.1007/BF01195555. PMID 8315413. S2CID 45871975.
- ^ Adalbert R, Nógrádi A, Szabó A, Coleman MP (October 2006). «The slow Wallerian degeneration gene in vivo protects motor axons but not their cell bodies after avulsion and neonatal axotomy». The European Journal of Neuroscience. 24 (8): 2163–8. doi:10.1111/j.1460-9568.2006.05103.x. PMID 17074042. S2CID 25359698.
- ^ Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z (August 2005). «A local mechanism mediates NAD-dependent protection of axon degeneration». The Journal of Cell Biology. 170 (3): 349–55. doi:10.1083/jcb.200504028. PMC 2171458. PMID 16043516.
- ^ Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP (January 2007). «NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration». Cell Death and Differentiation. 14 (1): 116–27. doi:10.1038/sj.cdd.4401944. PMID 16645633.
- ^ Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP (October 2010). «Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo». The Journal of Neuroscience. 30 (40): 13291–304. doi:10.1523/JNEUROSCI.1189-10.2010. PMC 6634738. PMID 20926655.
- ^ Fujiki M, Zhang Z, Guth L, Steward O (July 1996). «Genetic influences on cellular reactions to spinal cord injury: activation of macrophages/microglia and astrocytes is delayed in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration». The Journal of Comparative Neurology. 371 (3): 469–84. doi:10.1002/(SICI)1096-9861(19960729)371:3<469::AID-CNE9>3.0.CO;2-0. PMID 8842900. S2CID 8797673.
- ^ Brown MC, Perry VH, Hunt SP, Lapper SR (March 1994). «Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration». The European Journal of Neuroscience. 6 (3): 420–8. doi:10.1111/j.1460-9568.1994.tb00285.x. PMID 8019679. S2CID 37501852.
- ^ Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR (July 2012). «dSarm/Sarm1 is required for activation of an injury-induced axon death pathway». Science. 337 (6093): 481–4. Bibcode:2012Sci…337..481O. doi:10.1126/science.1223899. PMC 5225956. PMID 22678360.
- ^
- ^ Lee HC, Zhao YJ (2019). «Resolving the topological enigma in Ca 2+ signaling by cyclic ADP-ribose and NAADP». Journal of Biological Chemistry. 294 (52): 19831–19843. doi:10.1074/jbc.REV119.009635. PMC 6937575. PMID 31672920.
- ^ a b Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J (April 2015). «SARM1 activation triggers axon degeneration locally via NAD⁺ destruction». Science. 348 (6233): 453–7. Bibcode:2015Sci…348..453G. doi:10.1126/science.1258366. PMC 4513950. PMID 25908823.
- ^ a b
- ^ Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J (October 2016). «+ depletion». eLife. 5. doi:10.7554/eLife.19749. PMC 5063586. PMID 27735788.
- ^ Gilley J, Ribchester RR, Coleman MP (October 2017). «S, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy». Cell Reports. 21 (1): 10–16. doi:10.1016/j.celrep.2017.09.027. PMC 5640801. PMID 28978465.
- ^ Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M (January 2015). «Pathological axonal death through a MAPK cascade that triggers a local energy deficit». Cell. 160 (1–2): 161–76. doi:10.1016/j.cell.2014.11.053. PMC 4306654. PMID 25594179.
- ^
- ^ Henninger N, et al. (2016). «Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1». Brain. 139 (4): 1094–1105. doi:10.1093/brain/aww001. PMC 5006226. PMID 26912636.
External links[edit]
- Wallerian+Degeneration at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
Focal nerve fiber degeneration at central fascicular or subperineurial regions is characteristic of acute ischemic nerve damage (Fig. 31.2), indicating that the distribution and type of fiber pathology suggest ischemia.
From: Handbook of Clinical Neurology, 2014
Autophagy in the Degeneration of Optic Nerve and Spinal Cord Axons
Jan Christoph Koch, … Paul Lingor, in Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging, 2016
Axonal Degeneration
Axonal degeneration is a prominent pathological feature in many neurological diseases (Lingor et al., 2012). In neurodegenerative diseases like Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS), the axon and the presynaptic terminals are the first neuronal compartments to be affected, often preceding the death of the neuronal somata by years (Burke and O’Malley, 2013; Fischer and Glass, 2007). In neuroinflammatory diseases, like multiple sclerosis, the primary pathological immune response targets glia cells and myelin sheaths. During the course of the disease, however, disease progression and symptomatic burden are mainly driven by the concomitant axonal degeneration (Ferguson et al., 1997). In neurotraumatic diseases like spinal cord injury (SCI), axons are acutely lesioned at a defined location resulting in a complete block of neuronal signal transmission distal from the lesion site and severe functional deficits (Liu et al., 1997). When axons are lesioned within the central nervous system, the deficits are mostly irreversible due to their limited regenerative capacity (Liu et al., 2011). Therefore, a better understanding of the pathomechanisms of axonal degeneration and development of potential therapeutic approaches are crucial for the majority of all neurological diseases.
Different forms of axonal degeneration have been described based on the spatial relation to the lesion site (proximal vs distal) and the time scale (acute vs chronic) (Lingor et al., 2012). They represent active self-destructing cellular processes involving a determined cascade of various molecular players and need to be discriminated from apoptosis, which is the death of the whole neuron including soma and all neurites (Raff et al., 2002).
After a focal traumatic lesion of an axon, for example, in SCI or optic nerve crush, the adjacent 400–600 µm of the axon on both sides of the lesion undergo a rapid disintegration that is termed as acute axonal degeneration (Kerschensteiner et al., 2005; Knoferle et al., 2010). On the molecular level, the initial axonal injury leads to a rapid calcium influx into the axon and a consecutive transient rise of the intraaxonal calcium concentration within seconds after the lesion. Blocking this initial calcium influx with calcium channel inhibitors can almost completely prevent the following axonal degeneration (Knoferle et al., 2010). Downstream of calcium, the calcium-sensitive protease calpain is activated, modulating a multitude of intraaxonal molecular targets (Kerschensteiner et al., 2005; Vosler et al., 2008; and own unpublished observations). This molecular cascade leads to a condensation and misalignment of neurofilaments followed by a fragmentation of microtubules that becomes visible already at 30 min after lesion at the ultrastructural level (Knoferle et al., 2010). Later, local swellings (axonal bulbs) appear along the axon. They are most probably related to local impairments of axonal transport. Within 6 h after the lesion, the axon on the proximal and distal 400–600 µm from the lesion site disintegrates. A prominent ultrastructural and molecular feature of acute axonal degeneration is the rapid local activation of autophagy, including an increase in numbers of autophagosomes and microtubule-associated protein light chain 3 (LC3) isoform II (Knoferle et al., 2010; Koch et al., 2010; Ribas et al., 2015). These phenomena will be discussed in detail later.
After the fast disintegration of the adjacent parts of the lesioned axon during acute axonal degeneration, the rest of the axon remains morphologically stable within the following hours. At 24–72 h after lesion, the distal part of the axon undergoes Wallerian degeneration (Conforti et al., 2014). This progressive fragmentation morphologically resembles the fragmentation seen in acute axonal degeneration (Kerschensteiner et al., 2005). It proceeds directionally along the axon and finally leads to a complete removal of the distal part of the axon (Conforti et al., 2014). The molecular machinery behind Wallerian degeneration is intrinsic to the axon, although macrophages and glia cells assist in the final removal of the axon fragments. A key molecule in Wallerian degeneration is nicotinamide mononucleotide adenylyltransferase (NMNAT) which is neuroprotective under physiological conditions but is not transported anymore along the axon after lesion, resulting in the demise of the distal axon (Conforti et al., 2014).
In neurodegenerative diseases like PD and ALS, the axons degenerate more slowly over longer time-periods. A prominent form of axonal degeneration in these diseases is the “dying back degeneration” (Cavanagh, 1964), which starts with a synaptic dysfunction and is then followed by a degeneration of the whole axon in a distal-to-proximal direction (Cavanagh, 1964).
In chronic inflammatory diseases, focal axonal degeneration has been described recently (Nikic et al., 2011). It is characterized by a focal axonal swelling constituted of accumulated organelles and dysmorphic mitochondria accompanied by an impairment of axonal transport. In contrast to acute and chronic axonal degeneration, focal axonal degeneration may represent a transient feature induced by the local action of immune cells. Thus, the focal swelling can either completely resolve or progress to a Wallerian-like fragmentation of the axon.
Although these forms of axonal degeneration differ with regards to morphological criteria and time kinetics, they share some general molecular mechanisms. First of all, the elevation of the axoplasmic calcium concentration either via influx from the extracellular space or from intracellular calcium stores plays an important role (Stirling and Stys, 2010). Calcium activates a number of calcium-dependent proteases including calpain and calcineurin, which then cleave and degrade cytoplasmic proteins (Vosler et al., 2008). Another central pathomechanism of axonal degeneration is a dysfunction of mitochondria, which accumulate along the axon (Kilinc et al., 2009). This leads to an insufficient energy supply and increased levels of reactive oxygen species and calcium. Moreover, impaired axonal transport (Millecamps and Julien, 2013), excessive kinase activation, and pathological aggregation of proteins have been implied in axonal degeneration (Lingor et al., 2012). Finally, we and others have shown that autophagy plays a major role in axonal degeneration (Knoferle et al., 2010; Koch et al., 2014; Ribas et al., 2015; Wang et al., 2006; Yang et al., 2013). Below, we will discuss the effects of autophagy in axonal degeneration in detail for the optic nerve and the spinal cord.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128054215000100
Amyloidosis and Neuropathy
ROBERT A. KYLE, … PETER J. DYCK, in Peripheral Neuropathy (Fourth Edition), 2005
Pathology.
Axonal degeneration, sometimes with predominant involvement of the small myelinated and unmyelinated fibers, is seen. In the presence of peripheral neuropathy, amyloid deposits, usually in globular or diffuse form, are found infiltrating epineural and endoneurial connective tissue. In addition, epineurial and endoneurial blood vessel walls are frequently thickened by amyloid deposits. There is a gross loss of myelin fibers, which often show signs of active axonal degeneration. Electron microscopy often reveals a marked loss of unmyelinated fibers. Teased fiber studies show a predominance of axonal degeneration.
In a study by Sommer and Schroder,191 in four of five cases of AL with polyneuropathy, the amyloid deposits reacted with antisera to κ or λ light chains. Electron microscopy revealed bundles of immunogold-labeled amyloid fibrils in coated and uncoated single membrane-bound vesicles of endoneurial macrophages. Schwann cells did not contain intracellular amyloid, but their processes were entangled in amyloid fibrils and their basement membranes were sometimes fused with fibrillar masses. The authors concluded that the monoclonal light chains precipitated and formed amyloid fibrils in the presence of, and presumably with the assistance of, endoneurial cells. They postulated that inefficiency of phagocytosis was one of the major factors in developing amyloidosis.191
In summary, amyloid infiltrates the proximal dorsal root and sympathetic ganglia early in the course of the disease. Amyloid also infiltrates the peripheral nerves, where it deposits between nerve fibers and compresses and distorts the nerves. The vessel lumens are frequently compromised by amyloid deposition. The nerve fibers develop axonal degeneration and, to a lesser extent, segmental demyelination.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780721694917501113
Peripheral Nervous System Toxicity Biomarkers
Tirupapuliyur V. Damodaran, in Biomarkers in Toxicology (Second Edition), 2019
Biomarkers of Axonal Injury
Axon degeneration is a hallmark consequence of chemical neurotoxicant exposure (e.g., acrylamide), mechanical trauma (e.g., nerve transection, spinal cord contusion), deficient perfusion (e.g., ischemia, hypoxia), and inherited neuropathies (e.g., infantile neuroaxonal dystrophy). Regardless of the initiating event, degeneration in the PNS and CNS progresses according to a characteristic sequence of morphological changes. These shared neuropathologic features suggest that subsequent degeneration, although induced by different injury modalities, might evolve via a common mechanism. Studies indicate that Ca2+ accumulation in injured axons has significant neuropathic implications and is a potentially unifying mechanistic event. It was proposed that diverse injury processes can lead to axon degeneration by an increase in intraaxonal Na+ in conjunction with a loss of K+ and axolemmal depolarization. A spontaneous mutation called WldS (slow Wallerian degeneration) showed us that Wallerian degeneration is not a passive disintegration of axon, when the axon is severed from the cell body, but an active process mediated by Nmnat protein (Mack et al., 2001). More recently, using a powerful forward genetics screen, a new pathway that involves Sarm protein has been found to prevent axonal degeneration after traumatic injury (Osterloh et al., 2012).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128146552000116
Tetraparesis, Hemiparesis, and Ataxia
Michael D. Lorenz BS, DVM, DACVIM, … Marc Kent DVM, BA, DACVIM, in Handbook of Veterinary Neurology (Fifth Edition), 2011
Central Axonopathy
Axonal degeneration results from disease within the neuronal cell body or within the axon itself. Lesion distributions are variable. Central axonopathy usually consists of bilateral and symmetrical degeneration of the axon and myelin and affects both sensory and motor tracts of the spinal cord, with the longest fibers being the most vulnerable.175 Breeds described include Jack Russell and smooth fox terriers,176-178 Scottish terrier,179 and Labrador retriever.180 Lesions typify diffuse loss of myelinated fibers in the cerebellar white matter and/or in the dorsal funiculi and pyramidal tracts in the spinal cord. Swollen axons may show neurofilament accumulations. In Brown Swiss cattle, at 5 months of age affected calves develop paraparesis and ataxia, and signs insidiously progress over months. Histopathology reveals diffuse axonal degeneration in the spinal cord, with spheroids (axonal swellings) present in some brainstem nuclei and in the cerebellar granular layer.181,182
Those that selectively involve long-tract axons in the CNS and PNS are described as in central-peripheral distal axonopathy. Central-peripheral axonopathy of young dogs have been described in the Ibizan hound,149 Alaskan husky,183 boxer,184-186 Pyrenean mountain dog,187 and New Zealand Huntaway.188 Canine degenerative myelopathy is a central-peripheral axonopathy of older-aged dogs (see Chapter 6).
Those limited to the PNS are neuropathies.149,189 Axonal degeneration or myelin degeneration may predominate. Sensorimotor neuropathy often shows degenerative changes in spinal ganglia or in the dorsal horn or ventral horn of the spinal cord.189,190
Multisystem neuronal degeneration involves axonopathy and neuronopathy throughout the CNS and may include neuropathy (see also Chapter 8). These have been described in the cairn terrier,191-193 cocker spaniel,194 golden retriever,195 and rottweiler.196
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9781437706512100074
Medical Diagnosis & Therapy
Leonard A Levin, in Glaucoma (Second Edition), 2015
Axonal Degeneration
Axonal degeneration after retinal ganglion cell injury takes place in two directions. The degeneration towards the cell is called retrograde degeneration, and the one away from the cell (and the site of injury) is called Wallerian or orthograde degeneration. It is important to realize that the processes of death of the cell body and degeneration of the axon are independent.118 Axonal responses can be divided into those associated with how the axon locally responds to injury; and those associated with degeneration of the proximal and distal healthy axon as a result of the injury.
Mechanisms by which the axon degenerates after direct injury include excess accumulation of Ca++ ions, activation of calpains, loss of the membrane potential, and several other processes. In healthy fibers, ATP-dependent pumps support homeostasis of ionic gradients. When energy supply is limited, either due to inadequate delivery or excessive utilization, ion gradients break down, unleashing a variety of cascades leading to Ca++ overload, activation of destructive enzymes, and local axonal dissolution.119
Mechanisms for degeneration of the healthy remaining axon after injury are different, with most research focusing on Wallerian degeneration, i.e. loss of the distal segment. Neurons have a self-destruct program in their axon,118 parallel to the suicide program for apoptosis of the cell body. The program for Wallerian degeneration is initiated when the axon is injured. There is a naturally occurring mutation in mice, Wallerian degeneration slow (WldS), which blocks this axonal degeneration program.120 Interestingly, the WldS mutation does not itself prevent the calcium influx associated with early stages of axonal degeneration.121 Studies of experimental glaucoma in these mutant animals demonstrate loss of the RGC but preservation of the axon.122 This is the exact opposite of what occurs when the apoptosis program is blocked in glaucoma, where the cell body is preserved but the distal axons are lost.6,123 Together, these data prove that the retinal ganglion cell body death and axonal degeneration programs are distinct.124
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780702051937000613
In Vivo Visualization of Single Axons and Synaptic Remodeling in Normal and Pathological Conditions
Federico W. Grillo, … Vincenzo De Paola, in Axons and Brain Architecture, 2016
11.9 Degeneration
Axon degeneration is a characteristic event that occurs in many neurodegenerative conditions including glaucoma, stroke, traumatic brain injury, and motor neuropathies. Axonal degeneration occurs in at least three phases—an acute and rapid degeneration phase on both sides of the lesion (Kerschensteiner et al., 2005; Knoferle et al., 2010), followed by a period of quiescence/latency, then rapid cytoskeletal disassembly, fragmentation and granular degeneration of the axon distal to the injury site. This latter phase of distinct morphological and molecular changes is known as Wallerian degeneration (Coleman and Freeman, 2010). These phases of degeneration occur to variable extents in different regions and axonal populations within the nervous system, and can vary significantly according to the trauma paradigm. In the spinal cord, Thy1-GFP dorsal column axons injured by a fine needle undergo acute axonal degeneration occurring on both sides of the lesion up to several hundred micrometers from the lesion site, whereas in laser ablated membrane-bound GFP cortical neurons rapid axonal degeneration only occurs within a few micrometers either side of the lesion site (Canty et al., 2013b), before stabilization of the axon on both sides is achieved, albeit temporarily. In this latter paradigm, owing to the shorter distances involved, the fragmentation kinetics of the degenerating axons can be monitored in their entirety. Our unpublished observations indicate that the kinetics of fragmentation are quite unique compared to those described for in other regions of the CNS (Canty et al., submitted).
Diffuse axonal injury typically occurs following rapid acceleration/deceleration and is reproduced in experimental settings using impact acceleration models, inertial acceleration models, and fluid percussion models. Axons are stretched without undergoing immediate transection, and all models result in characteristic cytoskeletal rearrangements and eventual secondary axotomy depending on the severity of the trauma. Much of our understanding of secondary axotomy comes from in vitro models of injured cortical axons with direct transection (Chuckowree and Vickers, 2003; Dickson et al., 2000), mechanical stretching (Iwata et al., 2004), physical oscillation of neurons (Nakayama et al., 2001), and transient pressurized fluid deflection of axons (Chung et al., 2005). Directly observing such changes in in vivo models of diffuse axonal injury is more problematic with resulting vascular pathology often making the visualization of single axons difficult. Diffusion tensor imaging has been successfully used to detect white matter deterioration in both mild and moderate–severe closed-skull brain injury models in the mouse (Bennett et al., 2012). Changes in axial and mean diffusivity are observed which correlate with postmortem histological analysis—silver staining in mild injury, and APP accumulations in axonal swellings following moderate–severe injury. Similarly, computed topography and magnetic resonance imaging of human patients after head injury cannot show axonal pathology in any detail.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128013939000116
Plasma Membrane Repair
Héctor R. QuintáFrancisco J. Barrantes, in Current Topics in Membranes, 2019
2.4 The axolemma subjected to mechanical trauma
Axonal degeneration plays a key physiological role in nervous system growth. During development axonal degeneration is a requirement for rewiring temporary pathways and establishing new neuronal cell connections. In a “Darwinian” survival of the fittest-like process, neurons compete for activity or target (e.g., attractive trophic factor) cues. Growth cone collapse and F-actin depolymerization are two early events of axonal degeneration, as observed in an experimental model using embryonic dorsal root ganglion explants to recapitulate developmental axonal pruning triggered by trophic factor withdrawal. Nerve growth factor-deprived root ganglion neurons undergo a typical degenerative process which culminates in axonal fragmentation and detachment about 24 h after growth factor withdrawal (Unsain et al., 2018).
Axonal degeneration also occurs in disease. Traumatic brain injury (TBI) and spinal cord injury (SCI) are two pathological nosological entities generated by mechanical trauma in which the axonal shaft is affected by transection, stretch and/or compression (Nehrt, Hamann, Ouyang, & Shi, 2010; Ouyang et al., 2010; Quinta, Pasquini, Rabinovich, & Pasquini, 2014; Shi & Pryor, 2000). These injuries produce a breach in axolemmal integrity that alters the physiological homeostasis. Most noticeable is the disruption of the intracellular environment, due to the increase in permeability to ions and molecules associated with loss of membrane integrity.
Upon axonal injury the neuronal cells disclose mechanisms to self-protect from axonal growth-cone collapse (Quinta et al., 2016), promoting effective repair mechanisms. As stated by Sandoval-Guzman and Currie, regardless of the regenerative outcome, the initial detection and response to an injury is to restore barrier function and dispose of pathogens (Sandoval-Guzman & Currie, 2018). Axonal regeneration is not an exception to this rule and is triggered by sequential steps: the initial step is the sealing of the membrane breach to allow the re-formation of a growth-cone. However, this self-defense mechanism is not completely successful. The archetypal example of unsuccessful axonal repair is the failure of spinal cord axonal tracts to self-repair after SCI, leading to permanent, often complete loss of motor function. Although the mechanisms to seal the axolemma membrane after injury may not achieve a significant regeneration, they are able to limit damage and avoid the progression of degeneration.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/S1063582319300316
Inflammation and Axon Degeneration
V. Hugh Perry M.A., D.Phil., in Multiple Sclerosis As A Neuronal Disease, 2005
1. Active Axon Degeneration
Axon degeneration accompanies an extraordinary diversity of injury to, and diseases of, the nervous system, and a favored model to study the mechanisms that underlie axon degeneration has been the study of degeneration of the distal segment of a transected axon, wallerian degeneration. The morphological events that appear in wallerian degeneration are found at the end stage of almost every form of axon degeneration, suggesting that there is a final common pathway in the degeneration process (Griffin et al., 1996). It was long thought that the axon degenerated as a consequence of separation from the cell body leading to a withering of the distal process, an essentially passive process (Finn et al., 2000). The degeneration of the axon was also linked in acute transection to an influx of calcium that activated calcium-activated proteases within the axoplasm, which in turn degraded the axon cytoskeletal elements (Schlaepfer and Hasler, 1979).
The serendipitous discovery of a strain of mouse carrying a mutation that dramatically slows wallerian degeneration (Lunn et al., 1989) has radically changed our views of how axons degenerate (Finn et al., 2000; Coleman and Perry, 2002). In wild-type mice, and indeed all mammals, the axon segment distal to an injury normally undergoes wallerian degeneration within a few days of transection. In the mutant Wlds mice the axons of both CNS and PNS neurons may survive for several weeks separated from the cell body (Fig. 3). This simple observation tells us that axon degeneration in wild-type axons must be an active autodestructive process akin to programmed-cell-death or apoptosis of the cell body of neurons and other cells. The mutation in these mice has now been identified, and the Wlds gene was found to be a chimeric protein formed from UbE4b, an ubiquitin ligase, and nicotinamide mononucleotide adenylyltransferase (Nmnat), an NAD synthesizing enzyme (Mack et al., 2001). The mechanism of action of this gene is not known, but it clearly implicates the ubiquitin-proteasome pathway in axon degeneration (see later).

Figure 3. The remarkable phenotype of the Wlds mouse (Lunn et al., 1989). Central panel (B) illustrates the appearance of the normal sciatic nerve in a 1-μm semithin section stained with toluidine blue. Panel (C) illustrates the appearance of the distal nerve of a wild-type mouse nerve undergoing wallerian degeneration 7 days after transection. Panel (A) shows the remarkable preservation of the distal axon segment 7 days after sciatic nerve transection in the Wlds mouse.
The programmed-cell-death-like process in axons shares some similarities with the apoptotic cascades described at the neuronal cell soma, but there are also notable differences. Transected neurites undergoing wallerian degeneration in vitro express phosphatidylserine residues on the plasma membrane as detected by Annexin V, undergo blebbing, and lose their mitochondrial potential before degeneration (Sievers et al., 2003). Manipulations that protect the cell bodies of some neurons from apoptosis, such as raising extracellular potassium or intracellular cyclic adenosine monophosphate, also protect transected neurites in vitro (Buckmaster et al, 1995). However, the degeneration of neurites in vitro is not protected by caspase inhibitors (Finn et al., 2000; Sievers et al, 2003), and axons in vivo are not protected by overexpression of Bcl-2 (Burne et al., 1996), although they are protected in Bax deficient mice (Dong et al 2003; J.W. McDonald, personal communication).
Thus, the “molecules of destruction” secreted by inflammatory cells in the MS lesion act on axons that contain the biochemical machinery that maintains the balance between life and death of the axon. These pathways act in an analogous fashion to, but distinct from, the pro-apoptic and anti-apoptotic pathways maintaining the balance between life and death of the cell soma. It is important to note that the protection of the axon in wallerian degeneration by the Wlds gene has now been shown to extend to protection of axons subjected to taxol toxicity (Wang et al. 2003), to protection of axons in a mouse model of motorneuron disease (Ferri et al., 2003), and protection of axons from the axon degeneration present in a dysmyelinating mutant, the Po knockout mouse (Samsam et al., 2003). A simple hypothesis is that secretory products of inflammatory cells, or contact with inflammatory cells, leads to local activation of these autodestruction pathways in the axon, and thus, a local axon transection.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780127387611500183
White Matter Structure
Julia M. Edgar, Ian R. Griffiths, in Diffusion MRI (Second Edition), 2014
7.5.2 Axonal Degeneration
Axonal degeneration can be triggered by a range of diverse insults (Coleman, 2005). For example, it occurs in specific diseases such as multiple sclerosis, HSP, motor neuron disease, and Alzheimer’s disease. Degeneration may also result from focal damage, for example, as a direct consequence of spinal cord injury or secondary to stroke. The morphological changes that ensue are also varied. The degeneration that affects the axon distal to focal damage is known as Wallerian degeneration. Wallerian degeneration leads to secondary degeneration of the associated myelin. However, while the axon degenerates relatively quickly, leaving behind a myelin “wall,” myelin debris can persist for many months or even years. In contrast to Wallerian degeneration, which is thought to progress anterogradely from the lesion site, distal or length-dependent degeneration, which is characteristic of the spastic paraplegias and a number of peripheral neuropathies, appears to begin in the distal most portion of the axon (furthest from the cell body) and spreads retrogradely (towards the cell body). Focal axonal swellings or spheroids are encountered in several neurodegenerative disorders, including multiple sclerosis, motor neuron disease, spastic paraplegia, and Alzheimer’s disease. Whereas these may represent the endbulbs of fully transected axons, they do not necessarily lead to irreversible damage or indicate that their associated axons are severed (Edgar et al., 2004; Adalbert et al., 2009; Nikic et al., 2011).
For reasons that are not understood, smaller CNS axons are often preferentially vulnerable to degeneration. For example, smaller axons are damaged in multiple sclerosis and the small axons of the corticospinal tract are susceptible in HSP and ALS (Ganter et al., 1999; DeLuca et al., 2004b). The preferential vulnerability of small fibers is also seen in animal models of human disease, for example in the EAE model of multiple sclerosis (Papadopoulos et al., 2006) and in the Plp1 gene knockout mouse model of SPG2 (Griffiths et al., 1998) (see Section 7.4.2.1).
Axonal pathology in multiple sclerosis was reported over a century ago by Charcot (reviewed by Bjartmar et al., 2003). However, it was largely neglected until relatively recently when the prevalence of swollen and transected axons in multiple sclerosis lesions was demonstrated (Ferguson et al., 1997; Trapp et al., 1998). It is now generally accepted that the irreversible neurological disability seen in chronic multiple sclerosis is related to cumulative axonal degeneration (Bjartmar and Trapp, 2001). The extent of axonal degeneration in multiple sclerosis and in HSP has been examined in detail in the spinal cord. Utilizing the well-defined anatomy of this CNS region, DeLuca et al. (2004a) quantified axonal densities in formalin-fixed, paraffin-embedded post-mortem tissue from adult human males and females, at all levels of the posterior sensory tracts and in the crossed corticospinal tracts, in the lateral columns of the spinal cord. Sections 10 μm thick were stained with Palmgren silver to demonstrate axons. In multiple sclerosis tissue, average axon loss in the corticospinal tract ranged from 15 to 33%, depending on the level of the tract analyzed (DeLuca et al., 2004a). In a separate study by Bjartmar et al. (2000), axonal loss in demyelinated lesions within the spinal cord ranged from 45 to 84% (and see Section 7.5.1). In adult HSP patients, fiber numbers in the crossed corticospinal tracts were reduced by between 51 and 69%, depending on the spinal cord level that was examined (DeLuca et al., 2004b). Such large fiber losses, however, were associated with much smaller reductions in tract area, with corresponding decreases of 12 and 41%, respectively (DeLuca et al., 2004b), demonstrating that tract atrophy is not a good indicator of the extent of axonal degeneration. This was ascribed to the increased gliotic reaction (hypertrophy of astrocytes and increased microglial densities) and edema that accompanies axonal degeneration. In multiple sclerosis tissue, the extent of tract atrophy correlated with disease duration, being greater with increasing time from onset, probably reflecting the reduction in inflammation and edema in chronic multiple sclerosis lesions (DeLuca et al., 2004a).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B978012396460100007X
Metal Neuropathy
ANTHONY J. WINDEBANK, in Peripheral Neuropathy (Fourth Edition), 2005
Cellular Pathology
Axonal degeneration is the major feature of altered peripheral nerve morphology in human arsenic intoxication. There are no modern autopsy reports of changes in the nervous system related to arsenic poisoning. The best older report is that of Erlicki and Rybalkin, who described the changes in a patient who died from pneumonia during recovery from arsenic poisoning.57 The clinical features were typical of a moderately severe arsenic neuropathy. At autopsy in this case, pathologic changes were found in the spinal cord and peripheral nerves. Anterior horn cells were decreased in number both in particular groups of the ventral gray matter and diffusely. The remaining cells had lost angular contours, were rounded, and often lacked processes. Cytoplasm contained granular yellow or yellow-brown pigment. This may reflect the presence of lipofuscin, an abnormality occurring in spinal cords unaffected by specific diseases. Nuclei were ragged and sometimes pale. In the cervical and lumbar enlargements of the spinal cord, there was no marked abnormality except that posterior and anterior columns were markedly thinned and had decreased numbers of myelinated fibers. The radial and peroneal nerves were evaluated. The majority of myelinated fibers were abnormal, but the morphologic changes were not described in detail. In the authors’ opinion, some fibers had remained without degeneration, some were degenerating, and others were regenerating.
There are a number of reports concerning biopsies in patients with arsenic neuropathy36,52,74,96,111,124,158 in which histologic, morphometric, electron microscopic, and teased fiber changes are described in the sural nerve or a branch of the superficial peroneal nerve. The changes reported have been consistent with axonal degeneration. Chhuttani and colleagues36 and Heyman and associates96 reported increased endoneurial cellularity and interstitial fibrous tissue with some increase in the thickness of the perineurium. All authors reported a decrease in the number of myelinated fibers, and Dyck and colleagues demonstrated that this occurs equally across the range of all fiber diameters.52 Several authors have suggested that, in the case of a single dose of arsenic, all fibers are at the same stage of degeneration into linear rows of myelin ovoids. However, LeQuesne and McLeod have shown in two cases, each with a documented single exposure, that fibers at all stages of axonal degeneration may be seen in the biopsy specimen.124 Segmental demyelination is either absent or present at a very low frequency.72 Occasional onion bulbs are seen, which traditionally have been thought to indicate a chronic process involving repeated episodes of demyelination and remyelination. Ohta has pointed out that, at the electron microscope level, the core of these onion bulbs appears to be a degenerated axon and myelin debris, suggesting that they may represent a Schwann cell response to axonal degeneration.158 This study also mentioned that unmyelinated fibers showed little change. Quantitative studies of unmyelinated fibers have not been performed. In tissue culture, arsenic appears to specifically affect axonal structure rather than producing demyelination.222
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780721694917501162
Российский государственный медицинский университет;
НИИ цереброваскулярной патологии и инсульта, Москва

Аксональные полинейропатии: патогенез и лечение
Как цитировать:
Ковражкина Е.А. Аксональные полинейропатии: патогенез и лечение. Журнал неврологии и психиатрии им. С.С. Корсакова.
2013;113(6):22‑25.
Kovrazhkina EA. Axonal polyneuropathies: pathogenesis and treatment. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2013;113(6):22‑25. (In Russ.)
Полинейропатия (ПНП) — заболевание периферической нервной системы, развивающееся в результате диффузного поражения периферических нервов — их аксонов (аксональные полинейропатии), миелиновой оболочки (демиелинизирующие полинейропатии) либо тел нейронов (нейронопатии) [9]. В основе патогенеза полинейропатий аксонального типа лежит генерализованное повреждение осевых цилиндров периферических нервов.
Патогенез. Аксональная дегенерация (аксонопатия) — результат нарушения метаболизма нейрона вследствие недостаточной выработки энергии в митохондриях и/или нарушения аксонального транспорта. Миелиновая оболочка при аксонопатиях страдает вторично (вторичная демилинизация). Миелин может повреждаться в результате ишемии нервов (поражения vasa nevrorum), отложения токсичных для нерва веществ или иммунных комплексов в эндоневрии (что характерно, например, для сахарного диабета, особенно 1-го типа — с высокой гипергликемией и аутоиммунными нарушениями) [4]. Соответственно, среди причин аксонопатии периферических нервов указывают нарушения метаболизма, токсические влияния, ишемию нервных стволов, наследственную предрасположенность и аутоиммунные механизмы [5]. По этиологическому фактору подавляющее большинство аксональных ПНП составляют метаболические и токсические, среди которых первое место занимают диабетическая (до 50% среди пациентов с сахарным диабетом и до 90% поражений нервной системы при диабете [9, 12]) и алкогольная (до 50% среди больных алкоголизмом [2, 9]).
Причиной токсико-метаболических ПНП является экзо- и эндогенные интоксикации. Поступающие извне вещества или собственные его метаболиты, токсичные для периферических нервов, вызывают их повреждение. Тяжесть этого повреждения зависит от степени токсичности данного агента (выделяют острые метаболические ПНП, развивающиеся на фоне тяжелой общей интоксикации, например, ПНП при быстро нарастающих печеночной недостаточности или уремии, при отравлении фосфоорганическими веществами, мышьяком, свинцом, как нежелательный эффект лечения препаратами лития, цитостатиками), длительности его воздействия, собственных генетических особенностей метаболизма нервной ткани, немаловажную роль играет также аутоиммунный фактор [13].
Большинство часто встречающихся в клинике метаболических ПНП являются итогом длительного воздействия эндо- и экзогенного токсического агента (диабетическая, печеночная, уремическая, алкогольная, профессиональная, лекарственная). Результатом такой интоксикации является повреждение осевого цилиндра аксона. Клинически это проявляется не только нарушениями чувствительности и мышечной слабостью (что свойственно и для демиелинизирующих ПНП), но и мышечными гипотрофиями, выраженными трофическими нарушениями. Все эти признаки свидетельствуют о длительном страдании аксонов периферических нервов. При электронейромиографии (ЭНМГ) выявляется снижение (иногда — вплоть до полного отсутствия) амплитуды сенсорных потенциалов и М-ответов периферических нервов. У пациентов с ПНП аксонального типа зачастую в достаточной степени сохранены двигательные функции (отсутствуют выраженные парезы, больные сохраняют способность ходить, часто без дополнительной опоры), тогда как инвалидизирующими являются чувствительные (боли и парестезии) и трофические нарушения. Так, аксональная сенсомоторная ПНП является одним из важных патогенетических факторов развития синдрома диабетической стопы [3]. Аксональное повреждение нервов развивается медленно, исподволь, но при правильном лечении потенциально обратимо.
При массивном воздействии токсичного для периферической нервной системы агента, участии ишемического компонента (за счет страдания vasa nevrorum), аутоиммунных влияний развиваются ПНП аксонально-демиелинизирующего типа такие, например, как уремическая, свинцовая, амиодароновая, вызываемые воздействиями высокотоксичных для нервов веществами [9]. При наиболее распространенной диабетической ПНП, демиелинизирующий компонент максимально представлен при инсулинпотребном сахарном диабете (характеризующимся более высокой гипергликемией), а выраженность демиелинизации периферических нервов нарастает при резких повышениях уровня глюкозы крови [4, 7]. На представленность демиелинизирующего компонента при метаболических ПНП влияют наследственные факторы (генетически обусловленная миелинопатия, которая протекала бы субклинически без дополнительного воздействия токсического агента), аутоиммунное повреждение (например, миелинопатия более выражена при сахарном диабете 1-го типа, при котором иммунокомпетентные клетки повреждают ткань поджелудочной железы) [4, 14].
ПНП аксонально-демиелинизирующего типа протекают более тяжело, с выраженными парезами, сенситивной атаксией, нейропатическим болевым синдромом, но нередко, при своевременном устранении действия токсического фактора, быстро регрессируют. При очень тяжелом и упорном течении заболевания, учитывая роль аутоиммунного механизма в развитии данного типа метаболических ПНП, необходимо кратковременное назначение иммуномодулирующей терапии (чаще глюкокортикоидов, иногда цитостатиков) [9, 11].
Клиническое течение ПНП. Как уже было сказано, при длительном воздействии умеренно токсичного для нервов агента аксонопатия развивается медленно, что не всегда заметно. Преимущественное поражение бедно миелинизированых вегетативных и сенсорных волокон вызывает парестезии, повышенную холодовую чувствительность кистей и стоп, негрубые трофические нарушения. При ЭНМГ не всегда удается выявить характерные признаки сенсорной и моторной аксонопатии — снижение амплитуд сенсорных потенциалов и М-ответов — если исследуются крупные богато миелинизированные нервы, аксоны которых страдают позднее. Быстрее выявляются признаки вторичной миелинопатии, как клинические — присоединение нейропатических болей, парезов, так и миографические — снижение скорости распространения возбуждения (СРВ) по нервам.
При условии адекватной терапевтической стратегии — устранения действия повреждающего периферические нервы фактора, назначение препаратов, влияющих на метаболизм нервной ткани, — наступает ремиелинизация, что отражается и на ЭНМГ в виде нарастания СРВ, и клинически — в виде регресса парезов и нейропатических болей, улучшения чувствительности. Однако, если полинейропатия развивалась в условиях длительного действия токсического агента, поражение аксонов периферических нервов, снижение амплитуд сенсорных потенциалов и М-ответов на ЭНМГ сохраняется, поскольку осевые цилиндры нервов обладают гораздо меньшей способностью к регенерации, чем миелин.
Так, в проведенном исследовании [8], посвященном лечению алкогольной ПНП, ведущим ЭНМГ-признаком поражения периферических нервов у большинства пациентов имелось снижение СРВ. В условиях элиминации токсического фактора (в данном случае приема алкоголя) и лечения препаратом α-липоевой кислоты (берлитион в суточной дозе 600 мг), у пациентов быстро, в течение 1-го месяца, регрессировали ЭНМГ и клинические признаки миелинопатии — уменьшалась выраженность парезов и болевого синдрома (в баллах по визуальной аналоговой шкале с 5,2±1,0 до 2,6±0,5) достоверно (р<0,05) нарастала СРВ (по моторным волокнам n. tibialis с 36,93±1,12 до 42,22±0,8 м/с, по сенсорным волокнам n. suralis c 32,45±0,70 до 38,24±0,50 м/с), но признаки аксонопатии (снижение амплитуд сенсорных и моторных ответов периферических нервов, сенсорные выпадения по полиневритическому типу, трофические нарушения) сохранялись и после 6 нед лечения.
Таким образом, можно представить следующую схему развития и течения метаболических ПНП (см. таблицу): высокотоксичные, высококонцентрированные токсические агенты приводят к развитию полинейропатии аксонально-демиелинизирующего типа (в развитии которых также значительную роль играют наследственный, сосудистый, аутоиммунный факторы), длительно действующие эндо- и экзогенные яды приводят преимущественно к страданию аксонов с умеренно выраженным вторичным поражением миелина (так как постоянно идет процесс ремиелинизации) [7].
Лечение. Аксональное повреждение нервов как первичного, так и вторичного характера, обратимо за счет регенерации поврежденных аксонов и концевого спрутинга сохранившихся аксонов, однако этот процесс протекает медленно (месяцы), часто аксональная регенерация бывает неполной. Для регресса аксонопатии периферических нервов необходимо нивелировать воздействие токсического агента (коррекция метаболических и алиментарных нарушений, детоксикация, влияние на аутоиммунные механизмы), большое значение имеет также патогенетическая терапия, направленная на восстановление нарушенного метаболизма аксонов (митохондриальные нарушения, повреждения, вызванные окислительным стрессом).
Таким образом, в патогенетическом лечении аксональных ПНП, в зависимости от их нозологического и клинико-электрофизиологического варианта, должны присутствовать детоксикация и коррекция метаболических и алиментарных нарушений; сосудистая терапия (дезагреганты, венотоники, пентоксифиллин, вазопростан); препараты, воздействующие на универсальные механизмы поражения аксонов (витамины и витаминоподобные препараты); при необходимости — воздействие на аутоиммунные компоненты (глюкокортикоиды, плазмаферез). В некоторых случаях, когда представленность миелинопатии в структуре поражения периферических нервов велика и трудно отличить токсико-метаболическую полинейропатию от воспалительной (например, синдром Гийена-Барре у пациента с алкоголизмом или дебют хронической воспалительной демиелинизирующей ПНП у больного с сахарным диабетом) необходимо проводить пробную иммуномодулирующую терапию, ее быстрая эффективность позволит высказаться в пользу превалирования у пациента аутоиммуного механизма поражения периферических нервов [7, 11].
В лечении всех метаболических ПНП необходимо устранить (по возможности) поражающий периферические нервы яд и использовать препараты, улучшающие метаболизм нервной ткани периферических нервов. Последние необходимо применять в течение длительного времени, так как первым эффектом данных препаратов будет ускорение ремиелинизации, но для восстановления самих аксонов — а данные ПНП являются аксональными — требуется значительно большее количество времени.
α-липоевая (тиоктовая) кислота — витаминоподобное вещество, эндогенно образующееся в организме, как кофермент участвует в окислительном декарбоксилировании α-кетокислот. Основная функция эндогенной липоевой кислоты в организме — участие в аэробном метаболизме продукта гликолиза пирувата. Тиоктовая кислота является коферментом в окислительном декарбоксилировании пировиноградной кислоты до ацетил-КоА и α-кетоглутаровой до сукцинил-КоА в цикле Кребса. Облегчая превращение молочной кислоты в пировиноградную с последующим декарбоксилированием последней, α-липоевая способствует ликвидации метаболического ацидоза [6]. Тиоктовая кислота обладает сложным комплексным действием: гипогликемическим, липотропным, гепатопротекторным, антиатеросклеротическим, является мощным антиоксидантом. Липоевая кислота может существовать в окисленной (-S-S-) и восстановленной (SH-)-формах, благодаря чему реализуются ее коферментные и антиоксидантные функции. Восстановленная форма, дигидролипоевая кислота, служит донором электронов для восстановления других антиоксидантов (витамины С, Е и глутатион), осуществляет рецикл витамина Е при его истощении. Дигидролипоат повышает интра- и экстрацеллюлярный уровни глутатиона — эндогенного антиоксиданта.
Эффект экзогенно вводимой α-липоевой кислоты в отношении полиневритического синдрома впервые обнаружен при сахарном диабете. Обусловленная сахарным диабетом гипергликемия приводит к отложению глюкозы на матричных протеинах кровеносных сосудов и образованию конечных продуктов прогрессирующего гликозилирования, в результате чего уменьшается эндоневральный кровоток, возникает эндоневральная ишемия. α-липоевая кислота приводит к снижению уровня глюкозы в крови и повышению содержания гликогена в печени, обладает гипогликемическим действием. На фоне воздействия препарата уменьшается выраженность сенсорных симптомов полинейропатии — боли, жжения, ощущения онемения и «ползания мурашек» в конечностях.
Применение α-липоевой кислоты оказывает положительное влияние на универсальные механизмы аксонального повреждения, такие как повреждающее действие окислительного стресса и митохондриальная дисфункция — за счет антиоксидантного действия, повышения содержания глутатиона. Энергокорригирующее действие α-липоевой кислоты, тропное именно к аксонам нервов, способствует, в конечном итоге, быстрейшей регенерации аксонов [18].
Препарат не только редуцирует проявления окислительного стресса, но и оказывает влияние на сосудистый компонент поражения периферических нервов, нормализуя эндоневральный кровоток (что имеет большое значения, например, при дибетической микроангиопатии) [16, 17]. Комплексный механизм действия α-липоевой кислоты объясняет ее эффективность в отношении всех аксональных ПНП, патогенез которых связан с токсико-дисметаболическим и сосудистым факторами. Так, эффективность тиоктовой кислоты показана при уремической и алкогольной ПНП, при поражении периферических нервов, индуцированном цитостатиками [15]. Для регенерации аксонов периферических нервов на фоне токсико-метаболических влияний важны также детоксикационный и гепатопротекторный эффекты α-липоевой кислоты. Положительный эффект препарата отмечен в отношении заболеваний печени, печеночной комы, некоторых интоксикаций, в том числе алкогольной [6, 10].
При значительно выраженной аксонопатии, учитывая медленную скорость регенерации аксонов, необходимо длительное применение достаточно высоких доз α-липоевой кислоты. Обычно ее суточная доза составляет 600 мг. Предпочтительнее начинать лечение с внутривенного капельного введения препарата — 600 мг (24 мл раствора) в разведении на 200 мл физиологического раствора, длительность инфузии составляет от 2 до 4 нед в зависимости от тяжести ПНП. В особо тяжелых случаях препарат вводят внутривенно капельно в дозе 1200 мг в сутки. После переходят на пероральный прием α-липоевой кислоты — в таблетках по 600 мг не менее 2 месяцев [1].
В соответствии с такой клинической потребностью — необходимостью длительного приема достаточно высоких доз α-липоевой кислоты, учитывая потенциально обратимый, но медленный характер аксональной регенерации — разработаны и фармацевтические формы препарата. Например, берлитион 300 и берлитион 600, производимые в виде раствора для внутривенных вливаний по 12 и 24 мл соответственно (для начального этапа лечения ПНП), так и таблеток по 300 и 600 мг (для продолжения терапии).
Соблюдение всех условий патогенетического лечения ПНП — нормализация гликемии, прекращение поступления экзогенного яда, детоксикация, коррекция аутоиммунных нарушений, длительное применение препаратов, нормализующих аксональный метаболизм, способствует постепенной регенерации аксонов, уменьшению выраженности чувствительных и двигательных нарушений, болевого синдрома, парестезий, парезов. Для уточнения динамики различных видов метаболических ПНП на фоне подобного терапевтического подхода необходимы дальнейшие исследования.
Активная дегенерация аксонов после перерезания или раздавливания нервного волокна
| Повреждение нерва | |
|---|---|
|
|
| Флуоресцентные микрофотографии (100x) валлеровской дегенерации в перерезанных и раздавленных периферических нервах. Левая колонка проксимальнее травмы, правая — дистальнее. A и B: 37 часов после резки. C и D: 40 часов после увлечения. E и F: 42 часа после резки. G и H: 44 часа после травмы. | |
| Специальность | Неврология |
Валлеровская дегенерация — это активный процесс дегенерации, который возникает, когда нервное волокно разрезано или раздавлено, а часть аксона дистальнее повреждения (т.е. дальше от тела клетки нейрона ) дегенерирует. Связанный с этим процесс отмирания или ретроградной дегенерации, известный как «валлеровская дегенерация», происходит при многих нейродегенеративных заболеваниях, особенно тех, при которых нарушен транспорт аксонов, таких как БАС и болезнь Альцгеймера.. Исследования первичной культуры показывают, что неспособность доставить достаточное количество необходимого аксонального белка NMNAT2 является ключевым исходным событием.
Валлеровская дегенерация происходит после повреждение аксонов как в периферической нервной системе (ПНС), так и в центральной нервной системе (ЦНС). Это происходит в отделе аксона дистальнее места повреждения и обычно начинается в течение 24–36 часов после поражения. До дегенерации дистальный отдел аксона имеет тенденцию оставаться электрически возбудимым. После травмы аксональный скелет распадается, и аксональная мембрана разрушается. Дегенерация аксонов сопровождается деградацией миелиновой оболочки и инфильтрацией макрофагами. Макрофаги, сопровождаемые шванновскими клетками, служат для очистки остатков от дегенерации.
Шванновские клетки реагируют на потерю аксонов экструзией своих миелиновых оболочек, подавлением генов миелина, дедифференцировкой и распространение. В конце концов они выстраиваются в трубочки (ленты Бюнгнера) и экспрессируют поверхностные молекулы, направляющие регенерирующие волокна. В течение 4 дней после травмы дистальный конец части нервного волокна, проксимальной к поражению, посылает отростки к этим трубкам, и эти отростки привлекаются факторами роста, продуцируемыми шванновскими клетками в трубках. Если росток достигает трубки, он прорастает в нее и продвигается примерно на 1 мм в день, в конечном итоге достигая и повторно иннервируя ткань-мишень. Если ростки не могут добраться до трубки, например, из-за слишком широкой щели или образования рубцовой ткани, хирургическое вмешательство может помочь направить ростки в трубки. Регенерация ПНС эффективна, с почти полным восстановлением в случае поражений, которые возникают вблизи дистального окончания нервного окончания. Однако в спинном мозге выздоровление практически не наблюдается. Одно важное отличие состоит в том, что в ЦНС, включая спинной мозг, миелиновые оболочки производятся олигодендроцитами, а не шванновскими клетками.
Содержание
- 1 История
- 2 Дегенерация аксонов
- 3 Клиренс миелина
- 3.1 Клиренс в ПНС
- 3.2 Клиренс в ЦНС
- 4 Регенерация
- 4.1 Шванновские клетки и эндоневральные фибробласты в PNS
- 5 Медленная валлеровская дегенерация
- 5.1 Эффекты мутации Wld
- 6 SARM1
- 7 См. Также
- 8 Ссылки
- 9 Внешние ссылки
История
Валлериан дегенерация названа в честь Августа Волни Уоллера. В 1850 году Уоллер экспериментировал с лягушками, перерезав им языкоглоточный и подъязычный нервы. Затем он осмотрел дистальные нервы от места повреждения, которые были отделены от своих клеточных тел в стволе мозга. Валлер описал распад миелина, который он назвал «мозговым веществом», на отдельные частицы различного размера. Дегенерирующие аксоны образовывали капли, которые можно было окрашивать, что позволяло изучать ход отдельных нервных волокон.
Дегенерация аксонов
Хотя большинство реакций на травмы включают передачу сигналов притока кальция, способствующую повторному закрытию оторванных частей, повреждения аксонов изначально приводят к острой дегенерации аксонов (AAD), которая является быстрое разделение проксимального (части, близкой к телу клетки) и дистального конца в течение 30 минут после травмы. После разделения на обоих концах формируются дистрофические луковичные структуры, и перерезанные мембраны герметизируются. В дистальном сегменте происходит короткая латентная фаза, в течение которой он остается электрически возбудимым и структурно неповрежденным. Дегенерация сопровождается набуханием аксолеммы и, в конечном итоге, образованием бусинчатых аксональных сфероидов. Этот процесс занимает около 24 часов в ПНС и дольше в ЦНС. Сигнальные пути, ведущие к дегенерации аксолеммы, в настоящее время плохо изучены. Однако исследования показали, что этот процесс AAD не зависит от кальция.
Гранулярная дезинтеграция аксонального цитоскелета и внутренних органелл происходит после деградации аксолеммы. Ранние изменения включают накопление митохондрий в паранодальных областях в месте повреждения. Эндоплазматический ретикулум деградирует, митохондрии набухают и в конечном итоге распадаются. Происходит деполимеризация микротрубочек, за которой вскоре следует деградация нейрофиламентов и других компонентов цитоскелета. Распад зависит от протеаз убиквитин и кальпаин (вызванный притоком иона кальция), что позволяет предположить, что дегенерация аксонов является активным процессом, а не пассивным, как ранее неправильно понималось. Таким образом, аксон подвергается полной фрагментации. Скорость деградации зависит от типа травмы и также медленнее в ЦНС, чем в ПНС. Другим фактором, влияющим на скорость деградации, является диаметр аксона: более крупным аксонам требуется больше времени для деградации цитоскелета и, следовательно, требуется больше времени для дегенерации.
Клиренс миелина
Миелин представляет собой фосфолипидную мембрану, которая оборачивается вокруг аксонов, обеспечивая им изоляцию. Он продуцируется клетками Шванна в ПНС и олигодендроцитами в ЦНС. Очистка от миелина — следующий шаг в дегенерации Валлера после дегенерации аксонов. Очистка от остатков миелина различна для ПНС и ЦНС. ПНС намного быстрее и эффективнее очищает миелиновые остатки по сравнению с ЦНС, и шванновские клетки являются основной причиной этого различия. Другой ключевой аспект — изменение проницаемости гемато-тканевого барьера в двух системах. В ПНС проницаемость увеличивается по всей дистальной культи, но нарушение барьера в ЦНС ограничивается только местом повреждения.
Клиренс в ПНС
Реакция шванновских клеток на повреждение аксона быстрый. Период времени до начала дегенерации аксонов. Нейрегулины, как полагают, ответственны за быструю активацию. Они активируют рецепторы ErbB2 в микроворсинках шванновских клеток, что приводит к активации митоген-активируемой протеинкиназы (MAPK). Хотя активность MAPK наблюдается, механизм восприятия повреждений шванновскими клетками еще предстоит полностью понять. Ощущение сопровождается снижением синтеза липидов миелина и в конечном итоге прекращается в течение 48 часов. Миелиновые оболочки сначала отделяются от аксонов в области резцов Шмидта-Лантермана, а затем быстро разрушаются и укорачиваются, образуя бусообразные структуры. Шванновские клетки продолжают очищать миелиновые остатки, разрушая свой собственный миелин, фагоцитоз внеклеточный миелин и привлекают макрофаги к миелиновым остаткам для дальнейшего фагоцитоза. Однако первые несколько дней макрофаги не притягиваются к региону; следовательно, до этого момента шванновские клетки играют основную роль в очистке миелина.
Шванновские клетки, как наблюдали, рекрутируют макрофаги путем высвобождения цитокинов и хемокинов после обнаружения повреждения аксонов. Рекрутирование макрофагов помогает улучшить скорость очистки от миелинового мусора. Резидентные макрофаги, присутствующие в нервах, выделяют дополнительные хемокины и цитокины, чтобы привлечь дополнительные макрофаги. Дегенерирующий нерв также производит хемотаксические молекулы макрофагов. Еще одним источником факторов рекрутирования макрофагов является сыворотка. Задержка рекрутирования макрофагов наблюдалась у мышей с дефицитом В-клеток, лишенных сывороточных антител. Эти сигнальные молекулы вместе вызывают приток макрофагов, пик которого приходится на третью неделю после травмы. В то время как клетки Шванна опосредуют начальную стадию очистки миелинового мусора, макрофаги приходят, чтобы завершить работу. Макрофагам способствуют опсонины, которые маркируют мусор для удаления. Три основные группы, обнаруженные в сыворотке, включают комплемент, пентраксины и антитела. Однако было показано, что только комплемент помогает в фагоцитозе миелинового дебриса.
Murinson et al. (2005) наблюдали, что немиелинизированные или миелинизированные шванновские клетки при контакте с поврежденным аксоном входят в клеточный цикл, что приводит к пролиферации. Наблюдаемая продолжительность деления шванновских клеток составляла приблизительно 3 дня после повреждения. Возможные источники сигнала пролиферации приписываются рецепторам ErbB2 и рецепторам ErbB3. Эта пролиферация может еще больше повысить скорость очистки миелина и играет важную роль в регенерации аксонов, наблюдаемой в ПНС. Клетки Шванна выделяют факторы роста, которые привлекают новые отростки аксонов, растущие из проксимальной культи после полной дегенерации поврежденной дистальной культи. Это приводит к возможной реиннервации клетки или органа-мишени. Однако реиннервация не обязательно идеальна, так как возможное введение в заблуждение происходит во время реиннервации проксимальных аксонов в клетки-мишени.
Клиренс в ЦНС
По сравнению со шванновскими клетками, олигодендроцитам для выживания необходимы сигналы аксонов. На стадиях своего развития олигодендроциты, которые не могут установить контакт с аксоном и получить сигналы аксонов, подвергаются апоптозу.
. Эксперименты по Валлеровской дегенерации показали, что при повреждении олигодендроциты либо подвергаются запрограммированной клеточной гибели, либо переходят в состояние покоя. Следовательно, в отличие от клеток Шванна, олигодендроциты не могут очистить миелиновые оболочки и их остатки. В экспериментах на крысах миелиновые оболочки были обнаружены на срок до 22 месяцев. Следовательно, скорость клиренса миелиновой оболочки в ЦНС очень медленная и, возможно, может быть причиной препятствий в регенерационных возможностях аксонов ЦНС, поскольку отсутствуют факторы роста, привлекающие проксимальные аксоны. Еще одна особенность, которая в конечном итоге приводит к образованию глиального рубца. Это еще больше снижает шансы на регенерацию и реиннервацию.
Олигодендроциты не могут задействовать макрофаги для удаления мусора. Поступление макрофагов в место повреждения ЦНС происходит очень медленно. В отличие от ПНС, микроглия играет жизненно важную роль в валлеровской дегенерации ЦНС. Однако их рекрутирование происходит медленнее по сравнению с рекрутированием макрофагов в PNS примерно на 3 дня. Кроме того, микроглия может быть активирована, но гипертрофирована и не может трансформироваться в полностью фагоцитарные клетки. Те микроглии, которые действительно трансформируются, эффективно очищают от мусора. Дифференцировать фагоцитарную микроглию можно путем тестирования экспрессии главного комплекса гистосовместимости (MHC) класса I и II во время дегенерации Валлерова. Скорость выведения микроглии очень медленная по сравнению с макрофагами. Возможный источник вариаций скорости клиренса может включать в себя отсутствие активности опсонина вокруг микроглии и отсутствие повышенной проницаемости гематоэнцефалического барьера. Сниженная проницаемость может еще больше препятствовать инфильтрации макрофагов в место повреждения.
Эти данные позволяют предположить, что задержка валлеровской дегенерации в ЦНС по сравнению с ПНС вызвана не задержкой дегенерации аксонов, а скорее, из-за разницы в скорости клиренса миелина в ЦНС и ПНС.
Регенерация
Регенерация следует за дегенерацией. Регенерация ПНС происходит быстро, что позволяет возобновлять рост до 1 миллиметра в день. Также могут потребоваться трансплантаты для соответствующей реиннервации. Он поддерживается клетками Шванна за счет высвобождения факторов роста. Регенерация ЦНС происходит намного медленнее и почти отсутствует у большинства позвоночных. Основной причиной этого может быть задержка очистки миелиновых остатков. Миелиновые остатки, присутствующие в ЦНС или ПНС, содержат несколько ингибирующих факторов. Продолжительное присутствие миелиновых остатков в ЦНС может препятствовать регенерации. Эксперимент, проведенный на тритонах, животных, которые обладают способностью к быстрой регенерации аксонов ЦНС, показал, что валлеровская дегенерация повреждения зрительного нерва занимает в среднем от 10 до 14 дней, что также свидетельствует о том, что медленный клиренс тормозит регенерацию.
Шванновские клетки и эндоневральные фибробласты в ПНС
В здоровых нервах фактор роста нервов (NGF) продуцируется в очень малых количествах. Однако при повреждении экспрессия мРНК NGF увеличивается в пять-семь раз в течение 14 дней. Нервные фибробласты и шванновские клетки играют важную роль в повышении экспрессии мРНК NGF. Макрофаги также стимулируют шванновские клетки и фибробласты продуцировать NGF через интерлейкин-1, полученный из макрофагов. Другие нейротрофические молекулы, продуцируемые вместе шванновскими клетками и фибробластами, включают нейротрофический фактор головного мозга, нейротрофический фактор глиальных клеток, цилиарный нейротрофический фактор, фактор ингибирования лейкемии, инсулиноподобный фактор роста и фактор роста фибробластов. Вместе эти факторы создают благоприятную среду для роста и регенерации аксонов. Помимо факторов роста, клетки Шванна также обеспечивают структурное руководство для дальнейшего усиления регенерации. Во время фазы пролиферации шванновские клетки начинают формировать линию клеток, называемую полосами Бангнера, внутри базальной ламинарной трубки. Было обнаружено, что аксоны регенерируют в тесной связи с этими клетками. Клетки Шванна активируют выработку молекулы адгезии на клеточной поверхности ниндзюрин, дополнительно способствуя росту. Эти клеточные линии направляют регенерацию аксона в правильном направлении. Возможным источником ошибки, которая может возникнуть в результате этого, является возможное несовпадение целевых ячеек, как обсуждалось ранее.
Из-за отсутствия таких благоприятных стимулирующих факторов в ЦНС, регенерация в ЦНС задерживается.
Медленная валлеровская дегенерация
Мыши, принадлежащие к штамму C57BL / Wld, задерживают валлеровскую дегенерацию и, таким образом, позволяют изучать роль различных типов клеток и лежащих в их основе клеточных и молекулярных процессов.. Современное понимание этого процесса стало возможным благодаря экспериментам на мышах линии Wld. Мутация впервые произошла у мышей в Harlan-Olac, лаборатории по производству животных, Соединенное Королевство. Мутация Wld представляет собой аутосомно-доминантную мутацию, встречающуюся в хромосоме 4 мыши. Мутация гена представляет собой тандемную тройную повторность 85 т.п.н., встречающуюся в природе. Мутированный участок содержит два связанных гена: никотинамидмононуклеотид аденлилтрансфераза 1 (Nmnat1) и фактор убиквитинирования e4b (Ube4b). Линкерная область, кодирующая 18 аминокислот, также является частью мутации. Было показано, что защитный эффект белка Wld обусловлен активным сайтом, синтезирующим НАД в области NMNAT1.
Хотя созданный белок локализуется в ядре и едва обнаруживается в аксонах, исследования показывают, что его защитный эффект является из-за его присутствия в аксональном и терминальном отделах. Защита, обеспечиваемая белком Wld, присуща нейронам, а не окружающим опорным клеткам, и только локально защищает аксон, что указывает на то, что внутриклеточный путь ответственен за опосредование валлеровской дегенерации.
Эффекты мутации Wld
Мутация не причиняет вреда мыши. Единственный известный эффект заключается в том, что валлеровская дегенерация задерживается в среднем до трех недель после повреждения нерва. Сначала предполагалось, что мутация Wld замедляет инфильтрацию макрофагов, но недавние исследования показывают, что мутация защищает аксоны, а не замедляет макрофаги. Процесс, с помощью которого достигается защита аксонов, плохо изучен. Однако исследования показывают, что мутация Wld приводит к повышенной активности NMNAT1, что приводит к усиленному синтезу NAD. Это, в свою очередь, активирует SIRT1-зависимый процесс в ядре, вызывая изменения в транскрипции генов. НАД сам по себе может обеспечивать дополнительную защиту аксонов за счет увеличения энергетических ресурсов аксона. Однако более поздние работы вызывают сомнения в том, что NMNAT1 или NAD могут заменять полноразмерный ген Wld. Эти авторы продемонстрировали методами in vitro и in vivo, что защитный эффект сверхэкспрессии NMNAT1 или добавления NAD не защищает аксоны от дегенерации. Однако более поздние исследования показали, что NMNAT1 является защитным в сочетании с нацеливающим на аксоны пептидом, предполагая, что ключом к защите, обеспечиваемой Wld, является комбинация активности NMNAT1 и аксональной локализации, обеспечиваемой N-концевым доменом химерного белка. 57>
Обеспеченная защита аксонов задерживает начало валлеровской дегенерации. Следовательно, активация шванновских клеток должна быть отложена, поскольку они не будут обнаруживать сигналы деградации аксонов от рецепторов ErbB2. В экспериментах на мышах с мутациями Wld инфильтрация макрофагов значительно задерживалась на срок от шести до восьми дней. Однако, как только деградация аксонов началась, дегенерация протекает нормально, и, в зависимости от нервной системы, деградация следует с вышеописанной скоростью. Возможные последствия этого позднего начала — более слабые регенеративные способности у мышей. Исследования показывают, что регенерация может быть нарушена у мышей Wld, но это, вероятно, является результатом того, что окружающая среда неблагоприятна для регенерации из-за продолжающегося существования недогенерированных дистальных волокон, тогда как обычно остатки очищаются, уступая место новому росту.
SARM1
Путь валлеровской дегенерации был дополнительно прояснен открытием, что стерильный альфа- и TIR-мотив, содержащий белок 1 (SARM1), играет центральную роль в пути валлеровской дегенерации. Ген был впервые идентифицирован при скрининге мутагенеза Drosophila melanogaster, и впоследствии нокауты его гомолога у мышей показали надежную защиту перерезанных аксонов, сравнимую с таковой у Wld.
SARM1 катализирует синтез и гидролиз из циклической ADP-рибозы (cADPR) от NAD до ADP-рибозы. Активация SARM1 локально запускает быстрый коллапс уровней NAD в дистальной части поврежденного аксона, который затем подвергается дегенерации. Позднее было показано, что этот коллапс уровней NAD происходит из-за того, что TIR-домен SARM1 обладает внутренней активностью расщепления NAD. Белок SARM1 имеет четыре домена, сигнал митохондриальной локализации, аутоингибиторную N-концевую область, состоящую из мотивов броненосца / HEAT, два стерильных альфа-мотива, ответственных за мультимеризацию, и С-конец рецептора Toll / интерлейкина-1, обладающий ферментативной активностью. Активации SARM1 достаточно, чтобы снизить уровень НАД и запустить путь валлеровской дегенерации.
Активность SARM1 помогает объяснить защитную природу фактора выживания NMNAT2, как было показано ферментами NMNAT для предотвращения опосредованного SARM1 истощения НАД. Эта взаимосвязь дополнительно подтверждается тем фактом, что мыши, лишенные NMNAT2, которые обычно нежизнеспособны, полностью спасаются путем делеции SARM1, в результате чего активность NMNAT2 находится выше SARM1. Другие пути передачи сигналов, способствующие дегенерации, такие как путь киназы MAP, были связаны с активацией SARM1. Было показано, что передача сигналов MAPK способствует потере NMNAT2, тем самым способствуя активации SARM1, хотя активация SARM1 также запускает каскад киназ MAP, что указывает на существование некоторой формы петли обратной связи. Одно из объяснений защитного эффекта мутации Wld заключается в том, что область NMNAT1, которая обычно локализована в соме, заменяет лабильный фактор выживания NMNAT2 для предотвращения активации SARM1, когда N-концевой участок Ube4 белка WldS локализует его в аксон. Тот факт, что повышенная выживаемость аксонов Wld происходит из-за более медленного оборота Wld по сравнению с NMNAT2, также помогает объяснить, почему нокаут SARM1 обеспечивает более длительную защиту, поскольку SARM1 будет полностью неактивным независимо от активности ингибитора, тогда как Wld в конечном итоге будет деградировать. Возможные последствия пути SARM1 в отношении здоровья человека могут быть обнаружены на моделях животных, которые демонстрируют черепно-мозговую травму, поскольку мыши, которые содержат делеции Sarm1 в дополнение к Wld, демонстрируют уменьшение повреждения аксонов после травмы. Специфические мутации в NMNAT2 связали механизм валлеровской дегенерации с двумя неврологическими заболеваниями.
См. Также
- Аксонотмезис
- Соединительная ткань в периферической нервной системе
- Диффузное повреждение аксонов
- Камеры пищеварения
- Повреждение нерва
- Нейрорегенерация
- Повреждение периферического нерва
- Первичное и вторичное повреждение головного мозга
- Классификация Седдона
- Исследование повреждений спинного мозга
Ссылки
Внешние ссылки
| Классификация | D
|
|---|
- Wallerian + Degeneration в Национальной медицинской библиотеке США Медицинские предметные рубрики (MeSH)