Introduction
Mental illnesses are common, debilitating, and at times fatal (Kessler et al., 2005). Yet we know little about the pathophysiology of many of these conditions and existing treatments are partially effective and cause many side effects. Therefore, there is a great need for better insights into the neurobiology of psychiatric conditions and, as a corollary, for new treatment targets. Many lines of evidence suggest that white matter (WM) abnormalities are associated with psychiatric conditions. In fact, WM abnormalities are common in bipolar disorder (Heng et al., 2010), major depressive disorder (Arnone et al., 2012), and even attention deficit and hyperactivity disorder (van Ewijk et al., 2012). But the most pronounced and widespread abnormalities have been reported in schizophrenia. Therefore, we will discuss schizophrenia as a paradigmatic psychiatric disorder. In this review, we will discuss some of the evidence for WM abnormalities in schizophrenia, highlight what it can and cannot tell us about the biology and discuss emerging alternative approaches to the problem.
Schizophrenia is typically diagnosed at a young age, is life-long, and is among the leading causes of disability among people aged 15–35 (Global Burden of Disease, 2006). The cost of the illness is high and the suffering of patients with SZ and that of their families is great, as poor medication compliance, high rates of substance use disorder comorbidity, and suicide rates near 10% take their toll (Kessler et al., 2005). Despite its significance, relatively little is known about the pathophysiology of schizophrenia. Several lines of evidence suggest that integration of activity across brain regions is as important as processing within any one brain region. These include deficits in integration of activity in large-scale neuronal networks (Garrity et al., 2007; Williamson, 2007; Whitfield-Gabrieli et al., 2009), abnormalities in WM integrity (Kubicki et al., 2007; Camchong et al., 2009), and in expression of myelin- and oligodendrocyte-related genes (Tkachev et al., 2003). WM abnormalities are critical to conceptualization of SZ as a dysconnection (abnormal connection) syndrome (Paus et al., 2008; Stephan et al., 2009).
MRI-Based Probes of White Matter Integrity
Although several MR parameters reflect tissue properties, the approach that is most widely used is diffusion MRI. As described elsewhere in this issue, water molecular diffusion, referring to the random translational (Brownian) motion of molecules, can be examined in vivo using diffusion MRI. The MRI signal decay when diffusion gradients are applied reflects the displacement distribution of water molecules. Because diffusion of water molecules is restricted by tissue components such as cell membranes or macromolecules, diffusion MRI provides unique information about the internal structure of brain tissue. In such experiments, a diffusion tensor is calculated and this consists of the three eigenvectors of diffusion arbitrarily labeled λ1, λ2, and λ3 from largest to smallest. Diffusion MRI has already been widely applied in the diagnosis and treatment of numerous brain disorders, most importantly in ischemic stroke where a fall in apparent diffusion coefficient [ADC; a.k.a. mean diffusivity = (λ1 + λ2 + λ3)/3] of water molecules is seen within hours of the ischemic event.
In the WM, water molecular diffusion takes place along the fiber orientation direction [λ// or axial diffusivity (AD) = λ1] to a much greater extent than perpendicular to it [λ⊥ or radial diffusivity (RD) = (λ2 + λ3)/2] (see Figure 1 for visual depiction of λ1, λ2, and λ3). This process can be measured using diffusion tensor imaging (DTI) where fractional anisotropy (FA—derived from λ1, λ2, and λ3) reflects coherence of diffusion. DTI has been used to demonstrate WM abnormalities in a variety of diseases including multiple sclerosis and schizophrenia. FA reductions are commonly interpreted as reflecting loss of “white matter integrity” but the exact nature of this loss cannot be determined using DTI because the abnormality could arise from intra- or extracellular water. In addition, there is exchange between the intra- and extracellular water compartments, making it impossible to deduce the biological source of any abnormalities. Therefore, reduced FA likely reflects different processes in different disorders (such as demyelination, fiber crossing, axonal swelling, or atrophy) (Alexander et al., 2007).
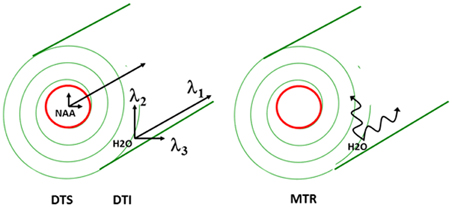
Figure 1. Schematic of proposed white matter measures.
Despite these limitations, DTI studies have provided strong evidence that WM integrity has great functional significance. For example, variation in DTI measures in healthy populations has been associated with cognitive processing speed (Turken et al., 2008). In schizophrenia, FA reductions have been associated with specific clinical presentations such as passivity phenomena (Sim et al., 2009), auditory hallucinations (Shergill et al., 2007), or positive symptoms more generally (Fujiwara et al., 2007), cognitive functioning including working memory (Kubicki et al., 2003; Karlsgodt et al., 2008), episodic memory (Nestor et al., 2004, 2008), executive function (Nestor et al., 2004, 2008; Rusch et al., 2007; Takei et al., 2009), verbal learning (Takei et al., 2008), and visuomotor performance (Perez-Iglesias et al., 2010), and with fMRI connectivity measures (Schlosser et al., 2007; Spoletini et al., 2009). Some similar findings have been reported in prodromal individuals as well (Koutsouleris et al., 2010). This literature suggests that WM integrity is highly relevant to specific domains of brain function and dysfunction. In its current state, however, the literature is weakened by the lack of a clear link between DTI and WM biology (Kubicki et al., 2007) as evidenced by the absence of a deeper biological understanding of WM abnormalities in schizophrenia and of novel treatment targets.
Novel Approaches
The limitations of DTI as currently implemented in most centers and for studying psychiatric conditions are widely acknowledged. There are multiple approaches for extracting additional information from the diffusion signal in order to generate novel biological insights for psychiatric research. In this review, we provide a selective review of a two-pronged approach that focuses on axon and myelin-related abnormalities separately. An extensive review of all possibilities is far beyond the scope of this review. Instead, we will primarily focus on the directions our research group is taking and introduce concepts that are useful for understanding other approaches and briefly describe some alternative approaches at the end of the paper.
The two-pronged approach utilizes two recently developed MR-based approaches to probe specific WM abnormalities: diffusion tensor spectroscopy (DTS), and magnetization transfer ratio (MTR). DTS is a diffusion MRI technique related to DTI which measures the diffusion of intracellular metabolites such as N-acetylaspartate (NAA) (see Figure 2). Because NAA is located exclusively in neurons and almost exclusively in the cytosol (as opposed to within organelles) (Tsai and Coyle, 1995), NAA diffusion provides information about neuronal microstructure. The ADC reflects the scalar distance traveled by a molecule in unit time, and it is the easiest measure to interpret. In DTI studies in schizophrenia, for example, water ADC elevations have been reported accompanying the widespread water FA reductions discussed above (DeLisi et al., 2006; Andreone et al., 2007; Nenadic et al., 2011). DTS measures have three very useful properties: first, although molecular diffusion can reflect either physical hindrance by membranes or cytosol “viscosity,” data collection parameters can be modified to ensure that we measure primarily the former (e.g., by increasing diffusion times and b-values). Second, diffusion measures are independent of metabolite concentration. Therefore, any metabolite concentration abnormalities do not confound measures of metabolite diffusion. Third, ADC is sensitive to axonal geometry and less to the macroscopic curvature of WM tracts. If a voxel is placed at an angle to fiber direction or if fibers zigzag instead of traveling straight, ADC will vary less than FA or RD. This is because ADC is an average of the three λ’s in the diffusion experiment and when fibers zigzag λ1 may go down but λ2 and λ3 rise, leaving their average approximately constant. In large voxel studies in vivo macroscopic curvature can often be a factor. Therefore, even though RD is intuitively more appropriate as an axon diameter measure, we focus on ADC. DTS approaches have been implemented and validated in previous work in a variety of contexts, including as probes of cellular diffusion (Ackerman and Neil, 2010) and for use in studies of axon diameter in the healthy WM (Upadhyay et al., 2008). The NAA DTS signal is informative: demyelination with preserved axon geometry would lead to no NAA ADC changes because NAA diffusion within axons would be unaffected. By contrast, axon diameter increase with preserved myelination would cause NAA ADC change (see Figure 3). It is important to note that the arguments presented above are relative, not absolute. NAA ADC can be impacted by multiple axon geometry factors such as macroscopic curvature and not just by axon diameter. While we propose that NAA ADC is useful in probing axonal geometry, it is only a partial index of axon diameter specifically. This is particularly true in brain regions such as the prefrontal cortex where there axons are not tightly packed.
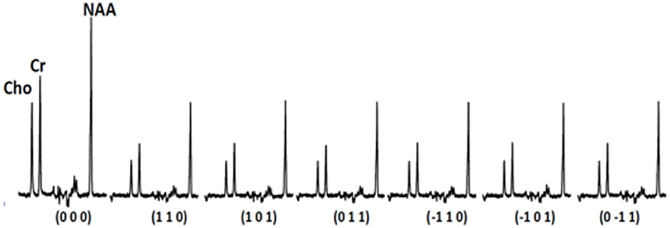
Figure 2. Sample spectra acquired during the diffusion experiment.
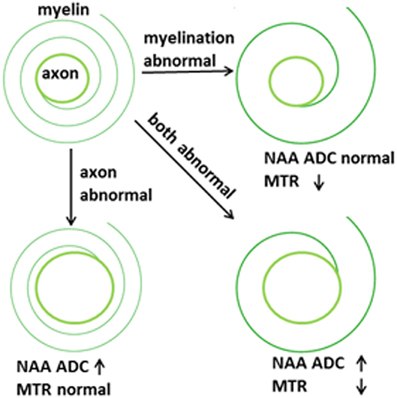
Figure 3. Proposed abnormalities in schizophrenia.
MTR is another MRI modality that has garnered recent attention. This approach relies on the exchange of magnetization between water molecules in different physical environments. In biological tissue, water molecules form a thin film around macromolecules, including myelin lipids. The “bound” water molecules in this film exchange protons with the “free” water molecules in cytosol and this exchange can be measured using a magnetization transfer paradigm where signal is saturated in one component (e.g., bound water) and the loss of saturation in the other component is measured (e.g., free water). The larger the amount of myelin complement in WM, the larger the proton exchange between bound and free water, and the higher the loss of signal from free water. This loss of signal is quantified as MTR. MTR imaging is robust enough to be it is used in clinical settings to improve contrast in WM anatomical imaging, especially in multiple sclerosis. MTR is reported to be reduced in schizophrenia, suggesting reduced myelin complement in this condition (Kubicki et al., 2005) although a recent study was discrepant (Mandl et al., 2008). The relationships between water DTI and NAA DTS signal and longitudinal vs. radial diffusion as well as between MTR and WM microstructure are summarized in the adjacent Figure 1.
Axon-Myelin Balance
Total WM volume is determined by the number of axons, their diameters, and the thickness of the myelin sheath around them. Although these parameters are inter-related, they are can vary partly independently. The importance of dissecting the processes that contribute to WM changes was highlighted recently in the context of normal human development. Increases in WM volume during adolescence have been traditionally interpreted as reflecting myelination but recent work using MTR showed that myelin content does not rise measurably in boys despite the fact that their WM volume increase is more pronounced than that of girls (Paus et al., 2008; Perrin et al., 2008, 2009; Paus, 2010). This pattern suggests that WM changes in male adolescence may be driven by increases in axonal diameter and number as opposed to an increase in the myelin complement.
The interplay between myelination and axon diameter is complex. Myelination speeds up conduction of action potentials. Larger axon diameters do the same, but the gain in conduction speed with growing axon diameters is less than that obtained from added myelination. Therefore, myelinated axons tend to be of small diameter to allow physical space for extra myelin. There appears to be a “sweet spot” for the ratio between axon diameter and fiber diameter (axon diameter + myelin sheath thickness) which maximizes conduction speeds. This is called the g-ratio and is calculated at around 0.6 for the human brain (Kandel et al., 1991; Chomiak and Hu, 2009; Paus and Toro, 2009). It is known that the g-ratio changes during brain development based on axonal electrical properties (Paus and Toro, 2009). In the extreme case, organisms with no myelin in the nervous system (such as the squid) have the largest axons, up to 1 mm in diameter (Chomiak and Hu, 2009; Paus and Toro, 2009). In pathological conditions where myelination is partial or degraded, axons can enlarge in diameter to compensate for reduced action potential speed (Nave, 2010).
The Challenge
Although DTI has been a very useful tool for psychiatric research, the ability to measure axon vs. myelin-related abnormalities separately in the WM is crucial for additional progress. DTI and MTR have been applied in schizophrenia, but there is no currently available measure that is axon-specific. A tool that provides axon specific along with myelin-related information would be valuable in identifying biologically meaningful abnormalities in this condition. MTR and DTS now provide this ability. In schizophrenia, there is strong evidence for myelination abnormalities (Hakak et al., 2001; Flynn et al., 2003; McCullumsmith et al., 2007; Uranova et al., 2007) as well as suggestions of a mechanistic relationship between developmental myelination abnormalities and schizophrenia (Budel et al., 2008). In addition, reductions in NAA levels have been reported in the WM in schizophrenia, suggesting a reduction in axonal packing density (Lim et al., 1998). Axonal diameter abnormalities have not been reported but until now these were only possible to measure in difficult postmortem electron microscopy studies. Axonal health and myelination are interrelated and abnormalities in one affect the other (Nave, 2010). Based on this literature we expect that application of DTS and MTR measures simultaneously will detect abnormalities in schizophrenia and in other psychiatric conditions. In this context, MTR functions as a marker of myelination and the NAA ADC as a marker of axonal geometry.
As reviewed above, correlation with cognitive and clinical outcomes have been used extensively to establish the pathophysiological significance of DTI abnormalities in schizophrenia. Armed with myelin- and axon-specific measures of WM integrity, it also becomes possible to explore the significance of each in similar fashion. For example, one would expect that cognitive tasks dependent on prefrontal circuitry may correlate with MTR and DTS measures obtained in the WM underlying the PFC. Another complementary approach to provide additional face validity for the combined MTR/DTS measures is the use of DTI to document abnormalities in water diffusion. The MTS/DTS approach can be used in conjunction with DTI (in fact, the water resonance measured during the DTS experiment is analogous to DTI data).
Taken together, the MTR/DTS approach would improve upon the existing paradigm of WM abnormalities in schizophrenia and other psychiatric conditions in two ways. First, it allows us to dissect the oft-repeated phrase “abnormal WM integrity” into component parts and examine how different changes in WM integrity can have different consequences. In fact, some WM alterations may be salutary ones as a compensation for upstream abnormalities. In this regard, the complex picture concerning WM changes during male vs. female adolescence discussed above provides a glimpse into how this proposal may challenge current paradigms of WM abnormality in psychiatry. Second, it offers concrete and measureable predictions about the impact of WM abnormalities on signal conduction in schizophrenia. I.e., “disrupted WM integrity” cannot correlate with conduction speed but axon geometry and myelin sheath thickness can. Although many have noted that WM abnormalities in schizophrenia must have functional consequences (Kubicki et al., 2007), the link between DTI measures and brain function remains abstract (Whitford et al., 2012).
Despite great interest in the role WM abnormalities play in cognitive function in psychiatric conditions, we do not yet have WM treatment targets. This is partly because existing measures of WM abnormalities are underdetermined, i.e., several mechanisms could lead to the same result. Therefore, we do not know which biological mechanisms are responsible for the WM alterations seen in schizophrenia. We propose that the MTR/DTS measure will offer meaningful biomarkers for this critical process.
There is a lengthy and productive history of studying postmortem tissue to probe WM abnormalities in psychiatry (e.g., Benes et al., 1987; Akbarian et al., 1993; Selemon and Goldman-Rakic, 1999), but in vivo neuroimaging tools have not been applied systematically to this problem except through the use of DTI. In addition, inherent problems with fixation often limit the application of electron microscopy to many postmortem human brain samples. Abnormalities in axons and in myelin have been reported, but we have had no ability to detect or monitor these in vivo. Although the application of MRI to histology has been discussed previously as “magnetic resonance microscopy” (Mori and Zhang, 2006; Mori et al., 2006), these ideas have not yet been expanded to human psychiatric disorders. Our vision is to expand MTR/DTS and related approaches into a “toolbox” measuring several aspects of the brain microenvironment in clinically acceptable scan times. To be maximally useful, this toolbox would collect regionally-specific data from voxels within gray matter (GM) and WM, and would include T1 and T2 relaxation times (Ongur et al., 2010) and perhaps other measures as well as MTR/DTS. Although this is a challenge with current technology, increasing field strengths and improving technical approaches are increasingly making it possible to use neuroimaging to probe parameters previously accessible only to postmortem research.
Magnetization Transfer Ratio (MTR) Spectroscopy
We have implemented the MTR sequence on the 4T Varian scanner at McLean Hospital and collected MTR data from a phantom preparation containing an aqueous NaCl-solution. The MTR experiment relies on measuring the total magnitude water signal in the presence and absence of a BISTRO saturation pulse. The water signal magnitude is maximal without the saturation pulse. The pulse causes saturation of signal coming from “bound” water molecules. Because there is transfer between “bound” and “free” water molecules, the saturation pulse measurably attenuates the signal coming from “free” water molecules. Figure 4 shows the MTR measured after a saturation pulse is applied at resonances near the main water resonance (normalized to a non-pulse intensity of 1.0). The lower curve represents measurements made in the aqueous solution phantom where there are no “bound” water molecules. When the saturation pulse is applied at most frequencies the MTR is zero, but it rises when the pulse is positioned right on the water frequency (i.e., it saturates all water molecule resonances). The data points from a healthy control collected from a 1 × 3 × 3 cm (9 cc) voxel in the WM underlying the right PFC show that, as expected, the saturation pulse causes a rise in MTR even when it is off-resonance in the human brain. This is because there are “bound” water molecules in vivo interacting with lipids and proteins and these are affected by the off-resonance saturation pulse. Their chemical exchange with the “free” water molecules causes a loss of water signal intensity. The MTR can be calculated based on water signal intensity acquired in presence (Ms) and absence (Mc) of the BISTRO saturation pulse [MTR = (Mc-Ms)/Mc].
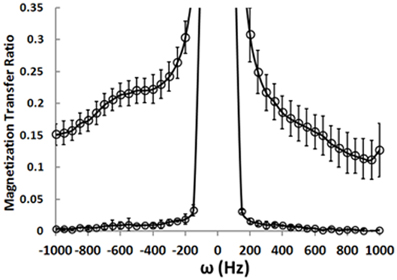
Figure 4. Magnetization transfer ratio data.
Compared to DTI, MTR is equally quick to obtain whole brain data. But the two techniques measure different processes, each with its own limitations. Therefore, the two techniques are best considered complementary.
DTS
We have also implemented DTS measurements of water and metabolites on the same 4T Varian scanner at McLean from the same 9 cc right PFC WM voxel in the human brain. DTS measurements in a phantom preparation demonstrated isotropic diffusion and these measures were used to calibrate diffusion gradient strength (b-value). The voxel location is identical to that in the MTR experiments. The seven spectra obtained during a DTS experiment are shown in Figure 2 with the NAA peak highlighted in select spectra. Note that we show water-suppressed spectra here for simplicity, although we also collected water unsuppressed spectra which allow us to calculate water ADC and FA. From these spectra are calculated the three diffusion eigenvalues and subsequently the ADC, RD, AD, and FA. The units for the diffusion data are mm2/s × 10−3. The water FA and ADC and the NAA FA and ADC values we calculate for a healthy control are similar to those in other DTI and DTS studies (e.g., Upadhyay et al., 2008; Camchong et al., 2009). Specifically, NAA is a larger molecule and diffuses more slowly than water so it has a lower ADC. Furthermore, two observations confirm that NAA has the diffusion properties of an intracellular metabolite while water does not: NAA FA is much higher than water FA; and the NAA AD/RD ratio is higher than the water AD/RD ratio. Being restricted within axons would increase FA and the AD/RD ratio, exactly as seen with NAA. The findings for creatine and choline (two other intracellular metabolites which are quantified with DTS) were similar to those for NAA. The metabolite diffusion parameters measured in a sample of 10 healthy individuals are listed in Table 1 below. These studies showcase our ability to implement a cutting-edge MRI sequence at 4T and to collect data on WM microstructure.
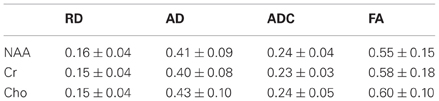
Table 1. Diffusion data in the healthy human white matter.
Compared to DTI, DTS collects data more slowly and from a single large voxel (as opposed to small voxels with whole brain coverage in DTI). These shortcomings are balanced by the different information offered by DTS—the diffusion properties of NAA are dependent on axon geometry and this information is complementary to the water diffusion information garnered using DTI.
Alternative Approaches
Some shortcomings of the single-voxel MTR/DTS approach would be remedied if we could collect data from the entire brain as is done in DTI studies. This is possible for MTR but not for DTS currently because metabolites such as NAA are present in the brain at about 1:5000 the concentration of water (approximately 10 mM vs. 50 M). This dramatically reduced signal forces us to collect data from a larger voxel and to carry out more repetitions for each data point to reach acceptable signal-to-noise ratios. Chemical shift imaging approaches have been described which can collect high quality MRS data from the entire brain (Posse et al., 2007; Maudsley et al., 2009) but these have not yet been combined with diffusion gradients. We plan to explore this combination but we recognize that this may not be possible given the SNR limitations.
Another concern with our DTS approach is that the NAA signal we measure in our experiments contains contributions from NAA and N-acetylaspartylglutamate (NAAG). NAAG is located both intra- and extracellularly (Coyle, 1997) and our DTS measures may be confounded by this contamination. NAAG is very similar to NAA in chemical structure so the two MRS signals are challenging to resolve using regular PRESS approaches. NAAG concentration in the PFC WM is 1.5 mM in healthy individuals (of which an unknown fraction is extracellular) (Pouwels and Frahm, 1997) whereas NAA concentrations are usually calculated at about 10 mM (Govindaraju et al., 2000). Therefore, we do not expect this to be a major factor in our work.
Finally MTR, although sensitive, is not a specific measure of myelin content. Concerns have been raised that MTR abnormalities can arise from acute inflammation, edema, and other processes that impact brain water content (Laule et al., 2007). This issue limits the utility of MTR in pathologies where gross abnormalities in brain water content are seen. There is no evidence for such gross abnormalities in psychiatric disorders and past applications of MTR in schizophrenia have been consistent with subtle myelination-related changes (Kubicki et al., 2005). Others have proposed a more specific measure of WM myelin content which takes advantage of the differential T2 relaxation properties of water trapped within myelin vs. free water (MacKay et al., 2006). This approach may be desirable because it would be more specific to myelin content, but it is more technically challenging to implement. The T2-based approach requires using ultrashort echo times (5 ms or shorter) which in turn require optimization of both hardware (amplifiers, transmit-receive switches) and software (e.g., STEAM sequences as opposed to our usual PRESS sequences).
There are also alternatives to the DTS approach, e.g., by optimizing the water diffusion experiment parameters for measuring axon diameter. Since the water signal is so much larger, such data could theoretically be collected in a shorter time than DTS. One such approach (termed AxCaliber) is in fact in early stages of development but thus far it has only been validated in vitro (Assaf et al., 2008). Unfortunately AxCaliber requires sequences to be repeated over very long scan times—and as a result is not available yet for clinical studies. Other approaches are also being developed (Zhang et al., 2011; Dyrby et al., 2012). Thus, we believe the single voxel DTS approach currently represents the state-of-the-art for examining changes in axon geometry.
Conclusions
A better understanding of WM abnormalities would be critical for the development of better treatments for common psychiatric conditions. Although DTI has shown us that these conditions are characterized by reduced WM integrity, it cannot tell us whether the abnormality is in axons, myelin, or both. This information is relevant because signal transduction in the WM depends on the relative health of these two compartments. Recently developed techniques such as MTR and DTS allow us to dissect signal from these two compartments, which would in principle help us quantify their relative abnormalities in psychiatric conditions. Many challenges remain, foremost among them the limitation of DTS data collection to a single large voxel and the difficulty of interpreting DTS signal from such large voxels. But future clinical studies will give us more detailed information on WM abnormalities in schizophrenia and other conditions than we have previously had access to. In addition, the application of these methods can extend beyond psychiatric disorders to study of aging and gender comparisons which may have additional implications for cross sectional and longitudinal evaluations in psychiatric studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akbarian, S., Bunney, W. J., Potkin, S., Wigal, S., Hagman, J., Sandman, C., et al. (1993). Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch. Gen. Psychiatry 50, 169–177.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Andreone, N., Tansella, M., Cerini, R., Versace, A., Rambaldelli, G., Perlini, C., et al. (2007). Cortical white-matter microstructure in schizophrenia. Diffusion imaging study. Br. J. Psychiatry 191, 113–119.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Arnone, D., McIntosh, A. M., Ebmeier, K. P., Munafo, M. R., and Anderson, I. M. (2012). Magnetic resonance imaging studies in unipolar depression: systematic review and meta-regression analyses. Eur. Neuropsychopharmacol. 22, 1–16.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Budel, S., Padukkavidana, T., Liu, B. P., Feng, Z., Hu, F., Johnson, S., et al. (2008). Genetic variants of Nogo-66 receptor with possible association to schizophrenia block myelin inhibition of axon growth. J. Neurosci. 28, 13161–13172.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Camchong, J., Lim, K. O., Sponheim, S. R., and Macdonald, A. W. (2009). Frontal white matter integrity as an endophenotype for schizophrenia: diffusion tensor imaging in monozygotic twins and patients’ nonpsychotic relatives. Front. Hum. Neurosci. 3:35. doi: 10.3389/neuro.09.035.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
DeLisi, L. E., Szulc, K. U., Bertisch, H., Majcher, M., Brown, K., Bappal, A., et al. (2006). Early detection of schizophrenia by diffusion weighted imaging. Psychiatry Res. 148, 61–66.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Dyrby, T. B., Sogaard, L. V., Hall, M. G., Ptito, M., and Alexander, D. C. (2012). Contrast and stability of the axon diameter index from microstructure imaging with diffusion MRI. Magn. Reson. Med. doi: 10.1002/mrm.24501. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Flynn, S. W., Lang, D. J., Mackay, A. L., Goghari, V., Vavasour, I. M., Whittall, K. P., et al. (2003). Abnormalities of myelination in schizophrenia detected in vivo with MRI, and post-mortem with analysis of oligodendrocyte proteins. Mol. Psychiatry 8, 811–820.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Fujiwara, H., Namiki, C., Hirao, K., Miyata, J., Shimizu, M., Fukuyama, H., et al. (2007). Anterior and posterior cingulum abnormalities and their association with psychopathology in schizophrenia: a diffusion tensor imaging study. Schizophr. Res. 95, 215–222.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Garrity, A. G., Pearlson, G. D., McKiernan, K., Lloyd, D., Kiehl, K. A., and Calhoun, V. D. (2007). Aberrant “default mode” functional connectivity in schizophrenia. Am. J. Psychiatry 164, 450–457.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Hakak, Y., Walker, J. R., Li, C., Wong, W. H., Davis, K. L., Buxbaum, J. D., et al. (2001). Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 98, 4746–4751.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Kandel, E. R., Schwartz, J. H., and Jessell, T. M. (1991). Principles of Neural Science. 3rd Edn. New York, NY: Elsevier.
Karlsgodt, K. H., van Erp, T. G., Poldrack, R. A., Bearden, C. E., Nuechterlein, K. H., and Cannon, T. D. (2008). Diffusion tensor imaging of the superior longitudinal fasciculus and working memory in recent-onset schizophrenia. Biol. Psychiatry 63, 512–518.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Kessler, R. C., Chiu, W. T., Demler, O., Merikangas, K. R., and Walters, E. E. (2005). Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62, 617–627.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Koutsouleris, N., Patschurek-Kliche, K., Scheuerecker, J., Decker, P., Bottlender, R., Schmitt, G., et al. (2010). Neuroanatomical correlates of executive dysfunction in the at-risk mental state for psychosis. Schizophr. Res. 123, 160–174.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Kubicki, M., McCarley, R., Westin, C. F., Park, H. J., Maier, S., Kikinis, R., et al. (2007). A review of diffusion tensor imaging studies in schizophrenia. J. Psychiatr. Res. 41, 15–30.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Kubicki, M., Park, H., Westin, C. F., Nestor, P. G., Mulkern, R. V., Maier, S. E., et al. (2005). DTI and MTR abnormalities in schizophrenia: analysis of white matter integrity. Neuroimage 26, 1109–1118.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Kubicki, M., Westin, C. F., Nestor, P. G., Wible, C. G., Frumin, M., Maier, S. E., et al. (2003). Cingulate fasciculus integrity disruption in schizophrenia: a magnetic resonance diffusion tensor imaging study. Biol. Psychiatry 54, 1171–1180.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Laule, C., Vavasour, I. M., Kolind, S. H., Li, D. K., Traboulsee, T. L., Moore, G. R., et al. (2007). Magnetic resonance imaging of myelin. Neurotherapeutics 4, 460–484.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Lim, K. O., Adalsteinsson, E., Spielman, D., Sullivan, E. V., Rosenbloom, M. J., and Pfefferbaum, A. (1998). Proton magnetic resonance spectroscopic imaging of cortical gray and white matter in schizophrenia. Arch. Gen. Psychiatry 55, 346–352.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
MacKay, A., Laule, C., Vavasour, I., Bjarnason, T., Kolind, S., and Madler, B. (2006). Insights into brain microstructure from the T2 distribution. Magn. Reson. Imaging 24, 515–525.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Mandl, R. C., Schnack, H. G., Luigjes, J., van den Heuvel, M. P., Cahn, W., Kahn, R. S., et al. (2008). Tract-based analysis of magnetization transfer ratio and diffusion tensor imaging of the frontal and frontotemporal connections in schizophrenia. Schizophr. Bull. 36, 778–787.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Maudsley, A. A., Domenig, C., Govind, V., Darkazanli, A., Studholme, C., Arheart, K., et al. (2009). Mapping of brain metabolite distributions by volumetric proton MR spectroscopic imaging (MRSI). Magn. Reson. Med. 61, 548–559.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
McCullumsmith, R. E., Gupta, D., Beneyto, M., Kreger, E., Haroutunian, V., Davis, K. L., et al. (2007). Expression of transcripts for myelination-related genes in the anterior cingulate cortex in schizophrenia. Schizophr. Res. 90, 15–27.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Mori, S., Zhang, J., and Bulte, J. W. (2006). Magnetic resonance microscopy of mouse brain development. Methods Mol. Med. 124, 129–147.
Pubmed Abstract | Pubmed Full Text
Nenadic, I., Wagner, G., Gullmar, D., Schachtzabel, C., von Consbruch, K., Kohler, S., et al. (2011). ADC changes in schizophrenia: a diffusion-weighted imaging study. Eur. Arch. Psychiatry Clin. Neurosci. 261, 213–216.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Nestor, P. G., Kubicki, M., Gurrera, R. J., Niznikiewicz, M., Frumin, M., McCarley, R. W., et al. (2004). Neuropsychological correlates of diffusion tensor imaging in schizophrenia. Neuropsychology 18, 629–637.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Nestor, P. G., Kubicki, M., Niznikiewicz, M., Gurrera, R. J., McCarley, R. W., and Shenton, M. E. (2008). Neuropsychological disturbance in schizophrenia: a diffusion tensor imaging study. Neuropsychology 22, 246–254.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Ongur, D., Prescot, A. P., Jensen, J. E., Rouse, E. D., Cohen, B. M., Renshaw, P. F., et al. (2010). T2 relaxation time abnormalities in bipolar disorder and schizophrenia. Magn. Reson. Med. 63, 1–8.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Perez-Iglesias, R., Tordesillas-Gutierrez, D., McGuire, P. K., Barker, G. J., Roiz-Santianez, R., Mata, I., et al. (2010). White matter integrity and cognitive impairment in first-episode psychosis. Am. J. Psychiatry 167, 451–458.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Perrin, J. S., Herve, P. Y., Leonard, G., Perron, M., Pike, G. B., Pitiot, A., et al. (2008). Growth of white matter in the adolescent brain: role of testosterone and androgen receptor. J. Neurosci. 28, 9519–9524.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Perrin, J. S., Leonard, G., Perron, M., Pike, G. B., Pitiot, A., Richer, L., et al. (2009). Sex differences in the growth of white matter during adolescence. Neuroimage 45, 1055–1066.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Posse, S., Otazo, R., Caprihan, A., Bustillo, J., Chen, H., Henry, P. G., et al. (2007). Proton echo-planar spectroscopic imaging of J-coupled resonances in human brain at 3 and 4 Tesla. Magn. Reson. Med. 58, 236–244.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Rusch, N., Spoletini, I., Wilke, M., Bria, P., Di Paola, M., Di Iulio, F., et al. (2007). Prefrontal-thalamic-cerebellar gray matter networks and executive functioning in schizophrenia. Schizophr. Res. 93, 79–89.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Schlosser, R. G., Nenadic, I., Wagner, G., Gullmar, D., von Consbruch, K., Kohler, S., et al. (2007). White matter abnormalities and brain activation in schizophrenia: a combined DTI and fMRI study. Schizophr. Res. 89, 1–11.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Selemon, L., and Goldman-Rakic, P. (1999). The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol. Psychiatry 45, 17–25.
Pubmed Abstract | Pubmed Full Text
Shergill, S. S., Kanaan, R. A., Chitnis, X. A., O’Daly, O., Jones, D. K., Frangou, S., et al. (2007). A diffusion tensor imaging study of fasciculi in schizophrenia. Am. J. Psychiatry 164, 467–473.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Sim, K., Yang, G. L., Loh, D., Poon, L. Y., Sitoh, Y. Y., Verma, S., et al. (2009). White matter abnormalities and neurocognitive deficits associated with the passivity phenomenon in schizophrenia: a diffusion tensor imaging study. Psychiatry Res. 172, 121–127.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Spoletini, I., Cherubini, A., Di Paola, M., Banfi, G., Rusch, N., Martinotti, G., et al. (2009). Reduced fronto-temporal connectivity is associated with frontal gray matter density reduction and neuropsychological deficit in schizophrenia. Schizophr. Res. 108, 57–68.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Takei, K., Yamasue, H., Abe, O., Yamada, H., Inoue, H., Suga, M., et al. (2008). Disrupted integrity of the fornix is associated with impaired memory organization in schizophrenia. Schizophr. Res. 103, 52–61.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Takei, K., Yamasue, H., Abe, O., Yamada, H., Inoue, H., Suga, M., et al. (2009). Structural disruption of the dorsal cingulum bundle is associated with impaired Stroop performance in patients with schizophrenia. Schizophr. Res. 114, 119–127.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Tkachev, D., Mimmack, M. L., Ryan, M. M., Wayland, M., Freeman, T., Jones, P. B., et al. (2003). Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362, 798–805.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Turken, A., Whitfield-Gabrieli, S., Bammer, R., Baldo, J. V., Dronkers, N. F., and Gabrieli, J. D. (2008). Cognitive processing speed and the structure of white matter pathways: convergent evidence from normal variation and lesion studies. Neuroimage 42, 1032–1044.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Upadhyay, J., Hallock, K., Ducros, M., Kim, D. S., and Ronen, I. (2008). Diffusion tensor spectroscopy and imaging of the arcuate fasciculus. Neuroimage 39, 1–9.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Uranova, N. A., Vostrikov, V. M., Vikhreva, O. V., Zimina, I. S., Kolomeets, N. S., and Orlovskaya, D. D. (2007). The role of oligodendrocyte pathology in schizophrenia. Int. J. Neuropsychopharmacol. 10, 537–545.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
van Ewijk, H., Heslenfeld, D. J., Zwiers, M. P., Buitelaar, J. K., and Oosterlaan, J. (2012). Diffusion tensor imaging in attention deficit/hyperactivity disorder: a systematic review and meta-analysis. Neurosci. Biobehav. Rev. 36, 1093–1106.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Whitfield-Gabrieli, S., Thermenos, H. W., Milanovic, S., Tsuang, M. T., Faraone, S. V., McCarley, R. W., et al. (2009). Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 106, 1279–1284.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Whitford, T. J., Ford, J. M., Mathalon, D. H., Kubicki, M., and Shenton, M. E. (2012). Schizophrenia, myelination, and delayed corollary discharges: a hypothesis. Schizophr. Bull. 38, 486–494.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Полинейропатия — это патология периферической нервной системы, которая развивается в результате диффузного повреждения периферических нервов и их аксонов. Отсюда и название болезни. В ее основе — генерализованное поражение осевого цилиндра периферических нервов.

Что такое аксональная полинейропатия
Полиневропатия (второе название — полиневрит) — это клинический синдром, который возникает из-за ряда факторов, влияющих на периферическую нервную систему, и отличается размытыми патогенетическими изменениями. Заболевание занимает одно из лидирующих мест в перечне недугов периферической нервной системы, уступая первенство только вертеброгенной патологии, превосходящей по сложности клинической картины и последствиям, развивающимся из-за нее.
Аскональная полинейропатия считается междисциплинарной проблемой, с ней часто сталкиваются доктора различных специализаций. В первую очередь с данным заболеванием обращаются к неврологу. Частота возникающего синдрома неизвестна, так как отсутствуют статистические данные.
На данный момент известны всего три важных патоморфологических механизма, которые лежат в истоках формирования полинейропатии:
- валлеровская дегенерация;
- первичная демиелинизация;
- первичная аксонопатия.
В соответствии с иммунологической теорией полинейропатия является результатом перекрестного образования иммунных глобулинов, уничтожающих собственные клетки, в результате чего возникает некроз тканей и мышечное воспаление.
Исследователи выдвигают ряд гипотез возникновения и проблем течения аксональной полинейропатии:
- Сосудистая. Базируется на вовлечении в процесс сосудов, по которым кислород и питательные вещества поступают в периферические нервы. Изменяются характеристики крови по качественному и количественному составу, что может привести к ишемии нервных окончаний.
- Теория оксидативного стресса. Позиционирует формирование болезни со стороны нарушения обмена оксида азота, вследствие чего изменяются калий-натриевые механизмы, лежащие в основе формирования нервного возбуждения и проведения импульсов по нервам.
- Теория деактивации факторов роста нерва. Говорит о том, что болезнь возникает из-за недостатка аксонального транспорта с последующим развитием аксонопатии.
- Иммунологическая. Объясняет развитие заболевания в результате перекрестного образования антител к структурам периферической нервной системы, которое сопровождается аутоиммунным воспалением, а затем и некрозом нервов.
Даже при использовании ультрасовременных методов диагностики сложно найти достоверную причину патологии, выяснить ее получается только у 50-70% пострадавших.
Факторов возникновения полинейропатии нижних конечностей по аксональному типу очень много. Однако даже инновационные способы исследования не позволяют установить истинную этиологию заболевания.
Мнение эксперта
Автор: Алексей Владимирович Васильев
Руководитель НПЦ болезни двигательного нейрона/БАС, кандидат медицинских наук, врач высшей категории
Аксональная полинейропатия — это одно из самых опасных неврологических заболеваний, сопровождающееся поражением периферической нервной системы. При болезни разрушаются периферические нервные волокна.
Причин возникновение аксональной полинейропатии несколько. Самые распространенные:
- Сахарный диабет нарушает структуру крови, питающей нервы, в свою очередь происходит сбой в обменных процессах.
- Длительный дефицит витаминов В. Именно они максимально важны для правильной работы нервной системы, поэтому долгая нехватка способна привести к аксональной полинейропатии.
- Воздействие токсинов на организм. К ним относят разнообразные отравляющие вещества, например, алкоголь, а также ВИЧ. При отравлении опасными веществами заболевание может развиться уже через несколько дней.
- Наследственный фактор.
- Синдром Гийена-Барре.
- Различные травмы, к которым также относится длительное сдавливание нервов, которое характерно при грыже или остеохондрозе.
Лечение аксональной полинейропатии обязательно должно быть комплексным, иначе нужного эффекта достичь не удастся. Категорически запрещается заниматься самолечением и при возникновении первых же симптомов нужно срочно обратиться к доктору. Врачи Юсуповской больницы подбирают лечение индивидуально для каждого пациента. В зависимости от тяжести патологии и симптоматики назначается комплексное лечение под наблюдением опытных специалистов.

Причины
Самые распространенные причины возникновения аксональной полинейропатии нижних конечностей:
- истощение организма;
- длительный недостаток витаминов группы В;
- недуги, ведущие к дистрофии;
- острые инфекции;
- токсическое поражение ртутью, свинцом, кадмием, угарным газом, спиртными напитками, метиловым спиртом, фосфорорганическими соединениями, медицинскими препаратами, принимаемыми без согласования с врачом;
- болезни сердечно-сосудистой, кроветворной, кровеносной и лимфатической систем;
- эндокринологические патологии, в том числе инсулинозависимость.
Главными факторами, которые провоцируют развитие моторной или сенсомоторной аксональной полинейропатии, являются:
- эндогенная интоксикация при почечной недостаточности;
- аутоиммунные процессы, протекающие в организме;
- амилоидоз;
- вдыхание токсических веществ или паров.
Также болезнь может быть обусловлена наследственностью.
Нехватка в организме витаминов группы В, а в особенности пиридоксина и цианокобаламина, крайне негативно воздействует на проводимость нервных и моторных волокон и может вызывать сенсорную аксональную полинейропатию нижних конечностей. Это же происходит при хронической алкогольной интоксикации, глистной инвазии, заболеваниях желудочно-кишечного тракта, которые ухудшают скорость всасывания.
Токсическое отравление лекарственными препаратами, аминогликозидами, золотыми солями и висмутом занимают большой процент в структуре факторов аксональной невропатии.
У пациентов с сахарным диабетом нарушена функция периферических нервов из-за нейротоксичности кетоновых тел, то есть метаболитов жирных кислот. Происходит это из-за невозможности организма использовать глюкозу как главный источник энергии. Поэтому вместо нее окисляются жиры.
При аутоиммунных заболеваниях, протекающих в организме, иммунная система человека атакует собственные нервные волокна, воспринимая их как источник опасности. Это происходит из-за провокации иммунитета, возникающей при неосторожном приеме иммуностимулирующих медикаментов и нетрадиционных методик лечения. Поэтому у людей, которые склонны к возникновению аутоиммунных заболеваний, пусковыми факторами аксональной полинейропатии являются:
- иммуностимуляторы;
- вакцины;
- аутогемотерапия.
При амилоидозе в организме накапливается такой белок, как амилоид. Именно он нарушает основные функции нервных волокон.
Первые признаки
Заболевание обычно начинает развиваться с поражения толстых или тонких нервных волокон. Зачастую аксональная полинейропатия имеет дистальное симметричное распределение на кисти или стопы. Нейропатия чаще всего сначала поражает нижние конечности, а затем симметрично распространяется вверх по телу. К самым частым первичным симптомам поражения относят:
- мышечную слабость;
- болевой синдром в конечностях;
- жжение;
- ощущение ползания мурашек;
- онемение кожных покровов.
Симптоматика ярче всего проявляется в вечернее и ночное время суток.
Симптомы
Врачи подразделяют хроническое, острое и подострое течение аксональной полинейропатии. Заболевание подразделяется на два вида: первично-аксональный и демиелинизирующий. В ходе течения болезни к ней присовокупляется демиелинизация, а затем и вторично аксональный компонент.
К основным проявлениям недуга относятся:
- вялость в мышцах ног или рук;
- спастический паралич конечностей;
- чувство подергивания в мышечных волокнах;
- головокружение при резкой перемене положения тела;
- отек конечностей;
- жжение;
- покалывание;
- ощущение ползания мурашек;
- снижение чувствительности кожных покровов к высокой или низкой температуре, боли и касаниям;
- нарушение ясности речи;
- проблемы с координацией.
Вегетативными признаками сенсомоторной полинейропатии асконального типа считаются следующие симптомы:
- учащенный или, напротив, замедленный сердечный ритм;
- неумеренное потоотделение;
- чрезмерная сухость кожи;
- изменение цвета кожных покровов;
- нарушение эякуляции;
- эректильная дисфункция;
- проблемы с мочеиспусканием;
- сбой двигательных функций желудочно-кишечного тракта;
- повышенное слюнотечение или, наоборот, сухость во рту;
- расстройство аккомодации глаза.
Заболевание проявляется в нарушениях функций поврежденных нервов. Именно периферические нервные волокна отвечают за двигательные функции мышечной ткани, чувствительность, а также оказывают вегетативное воздействие, то есть регулируют сосудистый тонус.
Для нарушения функции проводимости нервов характерны расстройства чувствительности, например:
- чувство ползания мурашек;
- гиперестезия, то есть увеличение чувствительности кожи к внешним раздражителям;
- гипестезия, то есть уменьшение чувствительности;
- отсутствие ощущения собственных конечностей.
Когда поражены вегетативные волокна, то из-под контроля выходит регуляция сосудистого тонуса. При аксонально-демиелинизирующей полинейропатии наступает сдавление капилляров, из-за чего ткани отекают. Нижние, а затем и верхние конечности из-за скапливания в них жидкости существенно увеличиваются в размерах. Так как при полинейропатии нижних конечностей основное количество крови накапливается именно в пораженных областях тела, то у пациента возникает стойкое головокружение при принятии вертикального положения. Из-за того, что пропадает трофическая функция, могут возникнуть эрозивно-язвенные поражения нижних конечностей.
Аксональная моторная полинейропатия проявляется в двигательных нарушениях верхних и нижних конечностей. Когда моторные волокна, отвечающие за движения рук и ног, повреждены, то наступает полный или частичный паралич мышц. Обездвиживание может проявляться совершенно нетипично — может ощущаться как скованность мышечных волокон, так и чрезмерная их расслабленность. При средней степени поражения ослаблен мышечный тонус.
В ходе течения заболевания могут быть усилены или ослаблены сухожильные и надкостничные рефлексы. В редких случаях доктор-невролог их не наблюдает. При болезни часто могут быть поражены черепные нервы, которые проявляются следующими нарушениями:
- глухотой;
- онемением подъязычных мышц и мускулатуры языка;
- невозможностью проглотить еду или жидкость из-за проблем с глотательным рефлексом.
Когда поражен тройничный, лицевой или глазодвигательный нерв, изменяется чувствительность кожных покровов, развиваются параличи, возникает асимметрия лица и подергивание мышц. Иногда при диагностированной аксонально-демиелинизирующей полинейропатии поражения верхних или нижних конечностей могут быть асимметричными. Такое случается при множественной мононейропатии, когда коленные, ахилловы и карпорадиальные рефлексы несимметричны.
Диагностика
Главной методикой исследования, которая позволяет обнаружить локализацию патологического процесса и степень пораженности нервов, является электронейромиография.
Чтобы определить причину заболевания, врачи назначают следующие анализы:
- определение уровня сахара в плазме крови;
- токсикологические тесты;
- полный анализ мочи и крови;
- выявление уровня холестерина в организме.
Нарушение нервных функций устанавливается при помощи определения температурной, вибрационной и тактильной чувствительности.
При первичном осмотре применяется зрительная методика исследования. То есть врач, к которому обратился с жалобами пострадавший, осматривает и анализирует такие внешние симптомы, как:
- уровень давления крови в верхних и нижних конечностях;
- чувствительность кожных покровов к прикосновениям и температуре;
- наличие всех необходимых рефлексов;
- диагностика отечности;
- изучение внешнего состояния кожи.
Выявить аксональную полинейропатию можно при помощи следующих инструментальных исследований:
- магнитная резонансная томография;
- биопсия нервных волокон;
- электронейромиография.
Лечение аксональной полинейропатии
Лечение аксональной полинейропатии должно быть комплексным и направленным на причину развития заболевания, его механизмы и симптоматику. Гарантией эффективной терапии является своевременное выявление болезни и лечение, которое сопровождается абсолютным отказом от сигарет, алкоголя и наркотических веществ, ведением здорового образа жизни и соблюдением всех рекомендаций врача. В первую очередь проводятся следующие терапевтические мероприятия:
- избавление от токсического воздействия на организм, если оно присутствует;
- антиоксидантная терапия;
- прием препаратов, которые воздействуют на тонус кровеносных сосудов;
- восполнение дефицита витаминов;
- регулярный контроль концентрации глюкозы в плазме крови.
Отдельное внимание уделяется лечению, направленному на купирование острого болевого синдрома.
Если присутствуют периферические парезы, то есть существенное снижение мышечной силы с многократным уменьшением амплитуды движений, то в обязательном порядке показана лечебная физкультура и специальные физические упражнения, направленные на возвращение тонуса мышечным тканям и предотвращение образования различных контрактур. Особенно важна регулярная психологическая поддержка, которая не дает пациенту впасть в депрессию, сопровождающуюся расстройством сна и чрезмерной нервной возбудимостью.
Лечение аксональной полинейропатии — это продолжительный процесс, так как нервные волокна восстанавливаются долго. Поэтому не стоит ожидать моментального выздоровления и возвращения к привычному образу жизни. Медикаментозная терапия включает такие препараты, как:
- обезболивающее;
- глюкокортикоиды;
- витамины группы В;
- антиоксиданты;
- сосудорасширяющие;
- средства, ускоряющие метаболизм и улучшающие микроциркуляцию крови.
Терапия лекарственными препаратами направлена на восстановление функций нервов, улучшение проводимости нервных волокон и скорости передачи сигналов центральной нервной системе.
Лечение следует проводить длительными курсами, которые не стоит прерывать, хоть и эффект от них проявляется не сразу. Чтобы устранить болевые ощущения и расстройство сна, назначают следующие медикаменты:
- антидепрессанты;
- противосудорожные;
- препараты, купирующие аритмию;
- обезболивающие.
Для избавления от боли используют нестероидные противовоспалительные препараты. Но стоит помнить, что применять их можно только короткий промежуток времени, так как длительное употребление может привести к повреждению слизистой оболочки желудочно-кишечного тракта.
К физиотерапевтическим методам лечения аксональной полинейропатии относятся:
- терапия магнитными волнами;
- грязелечение;
- электростимуляция;
- иглоукалывание;
- лечебный массаж;
- физкультура;
- ультрафонофорез;
- гальванотерапия.
Именно лечебная физкультура позволяет сохранить работоспособность мышечных тканей и поддерживать конечности в нужном положении. Регулярные занятия спортом вернут мышцам тонус, гибкость и увеличат амплитуду движений до нормальной.
Прогноз
Если заболевание обнаружено на ранней стадии и комплексно лечится квалифицированными специалистами, то прогноз для жизни и здоровья пациента более чем благоприятный. Стоит вести правильный образ жизни, рацион должен быть богат витаминами и минералами, необходимыми для правильного функционирования организма.
Если долгое время игнорировать болезнь и не предпринимать никаких действий, результат будет плачевным вплоть до полного паралича.
Профилактика
Пациент в обязательном порядке должен совершать профилактические мероприятия, которые помогут избежать рецидива или возникновения опасного заболевания. Они включают в себя обогащение рациона витаминами, регулярный контроль уровня сахара в крови, полный отказ от табакокурения, наркотических веществ и алкогольных напитков.
В целях профилактики болезни рекомендуется:
- носить удобную обувь, которая не пережимает стопу, ухудшая кровоток;
- регулярно осматривать обувь, чтобы избежать образования грибка;
- исключить пешие прогулки на длительные расстояния;
- не стоять долгое время на одном месте;
- мыть ноги прохладной водой или делать контрастные ванночки, что помогает улучшить циркуляцию крови в организме.
Пострадавшим в стадии ремиссии категорически запрещается принимать лекарственные препараты без согласования с лечащим врачом. Важно своевременно лечить воспалительные заболевания, соблюдать меры предосторожности при работе с токсическими веществами, которые оказывают пагубное воздействие на организм, регулярно выполнять лечебные физические упражнения.
Комплексная оценка функционального состояния центральной нервной системы в МЦ «CORTEX»
Человеческий мозг – сложная многоуровневая и многофункциональная система, работа которой является основой для нормальной деятельности всего организма. Оценка функционального состояния центральной и периферической нервной системы при различной патологии нервной системы является основой успешной реабилитации и формирования прогноза для каждого пациента.
В медицинском центре «Кортекс» проводится комплексная диагностика функционального состояния головного и спинного мозга пациентов, поступающих для проведения терапии. В центре используется новейшая диагностическая аппаратура научно-производственной фирмы «Нейрософт», позволяющая получать наиболее полную информацию о деятельности центральной нервной системы пациента: «Нейрон-Спектр 4/ВМП» и «Нейро –МВП» — многофункциональные компьютерные комплексы для проведения нейрофизиологических исследований. Интерпретация полученных данных проводится врачом функциональной диагностики высшей категории, кандидатом медицинских наук, доцентом кафедры нервных болезней медицинского университета.
Электроэнцефалография (ЭЭГ) — метод исследования функционального состояния головного мозга, основанный на регистрации его биоэлектрической активности через неповрежденные покровные ткани головы.
Современные электроэнцефалографы — это многоканальные приборы (чаще имеющие 8 или 16, иногда 20 и более усилительно-регистрирующих блоков — каналов), позволяющие одновременно регистрировать биотоки, отводимые от нескольких симметричных отделов головы. Исследование должно проводиться в свето- и звукоизолированном помещении.
На голову человека одевается специальная шапочка с электродами-антенами, соединенными с самим прибором. Сигналы, поступающие с коры головного мозга, передаются на электроэнцефалограф, который преобразует их в графическое изображение (волны). Это изображение напоминает ритм сердца на электрокардиограмме (ЭКГ).
Целью исследования является выявление эпилептической активности и определение типа эпилептических припадков; диагностика интракраниальных очагов поражения (абсцесс, опухоли); оценка электрической активности головного мозга при болезнях обмена веществ, ишемии мозга, его травмах, менингите, энцефалите, нарушении умственного развития, психических заболеваниях и лечении различными препаратами; оценка степени активности головного мозга, диагностика смерти мозга.
Электроэнцефалография применяется при всех неврологических, психических и речевых расстройствах. По данным ЭЭГ можно изучить цикл «сон и бодрствование», установить сторону поражения, расположение очага поражения, оценить эффективность проводимого лечения, наблюдать за динамикой реабилитационного процесса. Большое значение ЭЭГ имеет при исследовании больных с эпилепсией, поскольку лишь на электроэнцефалограмме можно выявить эпилептическую активность головного мозга.
Записанная кривая, отражающая характер биотоков мозга, называется электроэнцефалограммой (ЭЭГ). Электроэнцефалограмма отражает суммарную активность большого количества клеток мозга и состоит из многих компонентов. Анализ электроэнцефалограммы позволяет выявить на ней волны, различные по форме, постоянству, периодам колебаний и амплитуде (вольтажу). Электроэнцефалограмма (ЭЭГ) здорового человека имеет характерные черты: от всех областей коры отводится ритмическая активность с частотой около 10 Гц и амплитудой 50–100 мкВ — альфа-ритм. На электроэнцефалограмме (ЭЭГ) регистрируются также другие ритмы: как более низкие — дельта- и тета- (2–4, 5–7 Гц), так и более высокие— бета-ритмы (13–30 в сек), но амплитуда в норме их невысока и они перекрываются альфа-колебаниями.
В процессе перехода от младенчества к взрослому состоянию характер нормальной ЭЭГ постепенно меняется. В раннем детском возрасте на ней отражены главным образом медленные колебания, которые постепенно сменяются более частыми, и к 7 годам формируется альфа-ритм. Полностью процесс эволюции ЭЭГ завершается к 15—17 годам, приобретая к этому возрасту черты ЭЭГ взрослого человека. При значении патологической активности на ЭЭГ взрослого бодрствующего человека являются тета- и дельта-активность, а также эпилептическая активность.
Особенно значимым ЭЭГ-обследование оказывается при выявлении эпилептической активности, указывающей на предрасположенность к судорожным состояниям и проявляющейся следующими признаками:
- острые волны (пики) — колебание потенциала, имеющего крутое нарастание и крутой спад, острые волны могут быть единичными или групповыми, выявляются в одном или многих отведениях;
- комплексы пик—волна, представляющие собой колебания потенциала, состоящие из острой волны (пика) и сопутствующей ей медленной волны;
- пароксизмальные ритмы — ритмы колебаний в форме вспышек высокой амплитуды разной частоты, обычны пароксизмальные ритмы тета- и дельта-колебаний или медленных волн 0,5—1,0 Гц.
Расширению возможностей ЭЭГ в определении функционального состояния мозга и некоторых его патологических состояний, прежде всего эпилептической активности, способствуют специальные провокационные пробы: проба с гипервентиляцией — глубокие дыхательные движения с частотой 20 в минуту, ведущие к алкалозу и сужению сосудов мозга, проба со световым раздражителем — фотостимуляцией с помошью мощного источника света (стробоскопа), проба со звуковым раздражителем.
С помощью ЭЭГ получают информацию о функциональном состоянии мозга при разных уровнях сознания пациента. Достоинством этого метода являются его безвредность, безболезненность, неинвазивность, что явилось причиной широкого внедрения метода при диагностике и лечении различных заболеваний нервной системы.
Различные формы ДЦП и ЗПР часто сопровождаются наличием судорожных синдромов либо патологическими отклонениями на электроэнцефалограмме без судорог. Диагностика этих состояний ЦНС с помощью ЭЭГ является неотъемлемой частью стандарта обследования всех пациентов в МЦ «CORTEX», так как патологическая активность отдельных областей головного мозга или коры в целом резко тормозит дальнейшее развитие двигательных, речевых, когнитивных функций у больного ребенка и нуждается в лечении и постоянном динамическом наблюдении.
Вызванные потенциалы мозга — это электрическая активность головного мозга, возникающая на действие какого-либо стимула (звукового, зрительного, электрического). Деятельность анализаторов, осуществляющих взаимодействие человеческого организма с окружающей средой, подразумевает наличие специфического рецептора, воспринимающего раздражение, и поводящих путей, а также коркового представительства, на уровне которого осуществляется высшая функция анализа, синтеза и обратной связи. Рецепторы представляют собой минитрансформаторы, преобразующие энергию раздражителя в электрический потенциал, который может быть зафиксирован в виде потенциала действия любого звена трехнейронного сенсорного пути или в виде суммарного вызванного потенциала, отведенного со скальпа над конкретной областью коры головного мозга. Таким образом, поражение на любом уровне сенсорного пути неизбежно вызывает изменения характеристик вызванных потенциалов, вплоть до полного его отсутствия.
В зависимости от характера воздействующего стимула регистрируют вызванные потенциалы (ВП) мозга следующих модальностей:
- Слуховые (акустические стволовые) вызванные потенциалы – ответ на щелчок или тон
- Зрительные ВП – ответ на вспышку света, изображение предметов
- Соматосенсорные ВП – ответ на электрическую стимуляцию периферических нервов или тактильное раздражение
- Эндогенные, связанные с событиями (когнитивные ВП): с ожиданием, опозданием, принятием решения и инициацией двигательного ответа
- Вестибулярные миогенные – при выполнении отоневрологических тестов (раскручивание в специальном кресле).
В клинической практике применяются чаще ВП первых трех модальностей. Регистрация ВП мозга является объективным и неинвазивным, абсолютно безвредным методом исследования функций нервной системы. Исследования методики вызванных потенциалов является неоценимым средством раннего обнаружения и прогноза неврологических расстройств при различных заболеваниях головного и спинного мозга: инсульты, черепно-мозговая травма и травмы периферических нервов, рассеянный склероз, аномалии развития или опухоли головного и спинного мозга.
Основными целями регистрации ВП мозга являются:
- Выявление уровня поражения нервной системы;
- Определение распространенности процесса
- Определение характера поражения
- Определение степени тяжести патологического процесса
Исследование ВП мозга позволяет поставить диагноз, оценить прогноз и контролировать эффективность лечения.
Слуховые (акустические стволовые) вызванные потенциалы(СВП).
Метод отражает проведение слуховых импульсов от слухового нерва по стволовым и подкорковым структурам до коры головного мозга. Оценка СВП применяется для дифференциальной диагностики центральных и периферических поражений слуховоспринимающей системы, крайне полезны в ранней диагностике опухолей мосто-мозжечкового угла, еще при отсутствии клинических проявлений, а также применяются в диагностике нарушений речи и слуха у детей, при задержке речевого развития. В качестве стимула при исследовании СВП используются подаваемые через наушники звуковые стимулы – щелчки с интенсивностью звука около 100 Дб длительностью менее 1 мс и частотой стимуляции 10-15 Гц.
Показания к назначению СВП:
- Нарушение слуха, оценка нейросенсорной тугоухости
- Головокружение, нистагм, двоение в глазах, рассеянная легкая органическая симптоматика (косоглазие, асммметрия носогубных складок, мышечная дистония, нарушение координации движений, асимметрия рефлексов, патологические рефлексы, тошнота, головокружение, шум в ушах
- Дизартрия, дисфагия, заикание
- Оценка слуха у новорожденных
- Последствия родовых травм, в т.ч. перинатальной гипоксии и асфиксии
- Синдром внутричерепной гипертензии
Ранняя диагностика демиелинизирующих заболеваний (рассеянный склероз), опухоли ствола головного мозга.
В МЦ «CORTЕХ» исследование слуховых вызванных потенциалов проводится пациентам с ДЦП и ЗПР, имеющим нейросенсорную тугоухость, а также задержку речевого развития. У данной категории больных причиной задержки речевого и психического развития иногда является снижение слуха, а не поражение речевых центров. Методика СВП позволяет выявить патологию слуха у ребенка и дать соответствующие рекомендации по дальнейшему лечению.
Зрительные вызванные потенциалы мозга (ЗВП)
С помощью ЗВП можно получить объективную информацию о функциональном состоянии различных звеньев зрительного анализатора, выявить органические повреждения и определить уровень их локализации, определить наличие нарушений полей зрения. Регистрацию ЗВП необходимо проводить у больных при патологии зрительного нерва: невритах, демиелизирующих заболеваниях, для дифференцировки поражения на пре- и постхиазмальном уровне. Возможна объективизация состояния зрения у детей раннего возраста и в случае судебно-медицинской экспертизы, оценка зрительных нарушений и динамика при лечении. При регистрации короткие зрительные стимулы могут подаваться в виде вспышек, реверсии шахматных паттернов различного размера и т д. Фотостимуляция проводится через фотостимулятор, очки или экран монитора. Запись потенциалов сетчатки носит название нейроретинограмма.
Показания к обследованию:
- Снижение остроты зрения
- Травматическое повреждение зрительного нерва
- Атрофии зрительного нерва
- Токсическая невропатия
- Зрительные расстройства при нарушениях мозгового кровообращения
- Оценка зрения при зрительных агнозиях и повреждениях зрительной коры
- Оценка зрительных функций у больных с нарушениями сознания.
В нашем центре исследование зрительных потенциалов проводится детям, страдающим атрофией зрительного нерва, снижением зрения. Методика, проводимая в МЦ «CORTЕХ», позволяет оценить степень поражения зрительного нерва, а также отследить динамику проводимого лечения.
Соматосенсорные вызванные потенциалы мозга (ССВП )
Исследование проведения по чувствительным путям центральной нервной системы, в частности, по путям глубокой чувствительности, ответов центров спинного и головного мозга на электрическую стимуляцию периферических нервов. Практически можно зарегистрировать ВП афферентных волокон периферических нервов, проводящих путей и серого вещества спинного мозга, мозгового ствола и больших полушарий головного мозга, что является адекватной информацией о поражении как проводящих путей, так и сенсомоторной коры. В МЦ «CORTЕХ» исследование проводится с использованием многофункционального компьютерного комплекса для миографии и исследования вызванных потенциалов мозга — «Нейро –МВП».
Соматосенсорные вызванные потенциалы (ССВП) используются в диагностике различных демиелинизирующих, дегенеративных и сосудистых поражений центральной нервной системы. Помимо поражений головного мозга ССВП могут применяться как дополнительный метод в диагностике плексопатий и радикулопатий, в качестве подтверждающего теста используются при диабетической полинейропатии и др.
Для стимуляции чаще всего выбирают срединный нерв (верхние конечности) и большеберцовый нерв (нижние конечности). При наличии специальных показаний может производиться стимуляция других периферических нервов.
Регистрирующие электроды располагаются по ходу восходящих соматосенсорных путей – на уровнях периферических нервных сплетений, спинного и головного мозга. Количество электродов и уровней регистрации определяются клинической задачей. Подается около 500-1000 стимулов, ответы усредняются. Результат представляет последовательность колебаний, которые отражают прохождение нервных импульсов по восходящим путям, вплоть до сенсомоторной коры. Измеряются время и амплитуда каждого компонента, которые сравниваются затем с нормативными значениями.
Отсутствие или существенное снижение амплитуды компонента ВП говорит о наличии патологического процесса на уровне или ниже уровня его генерации. Увеличение латентности свидетельствует о замедлении проведения, вызванного, возможно, демиелинизирующим процессом.
Показания к обследованию:
- Травматическое поражение периферических нервов
- Поражение спинного мозга (определение уровня поражения и степени тяжести)
- Ранняя диагностика рассеянного склероза
- Нарушения чувствительности, онемения конечностей.
При различных формах детского церебрального паралича имеют место двигательные нарушения в виде парезов, параличей или атаксии. Исследование ССВП позволяет оценить состояние периферических нервов, степень их поражения, а также количество функционально активных нервных клеток в двигательных и чувствительных центрах спинного мозга, подкорковых структур и коры головного мозга, топическое расположение пораженного участка для более направленного и эффективного воздействия. Это позволит получить положительные эффекты от проводимой терапии в виде улучшения двигательной функции конечностей, улучшения ходьбы, увеличения силы ловкости в руках и ногах, развитие новых двигательных навыков у детей, страдающих ДЦП и ЗПР.
Когнитивные вызванные потенциалы( КВП)
Одним из методов, значительно продвинувших анализ и понимание процессов работы мозга, связанных с механизмом восприятия информации и ее обработки, является метод когнитивных вызванных потенциалов – КВП. Процессы узнавания и запоминания, а также принятия ответного решения сопровождаются более или менее закономерными нейродинамическими изменениями, которые можно объективно зафиксировать. В клинической практике применяется методика выделения когнитивных (связанных с процессами мышления) эндогенных ВП, обусловленных распознаванием и подсчетом слуховых стимулов (щелчков), отличающихся по частоте. Пациенту дается инструкция посчитать число «значимых» стимулов (щелчков с частотой тона 2000Гц и вероятностью подачи до 30%), не обращая внимания на «незначимые» (с частотой 1000 Гц и вероятностью подачи до 70 %). ВП на «незначимый» стимул представляет собой характерную V-волну. В ответ на «значимый» стимул заметен выраженный поздний позитивный компонент Р3 с латентностью около 300мс, чье наличие связывают с узнаванием, запоминанием и подсчетом стимулов. По характеристикам этой волны описанную методику часто называют Р 300. На параметры Р 300 влияют способность пациента к опознанию стимула и поддержанию внимания, уровень бодрствования, а также объем его оперативной памяти. в связи с этим методика используется для диагностики доклинических стадий деменции, оценки результатов лечения, побочного действия препаратов, а также при для профотбора.
Показания к обследованию:
- Нарушение памяти, внимания
- Ранняя диагностика когнитивных нарушений
- Оценка начальных когнитивных расстройств при хронической ишемии мозга, энцефалопатиях различного генеза, Паркинсонизме, эпилепсии и других заболеваниях
- Оценка динамики когнитивных нарушений в процессе лечения
- Оценка выраженности когнитивных нарушений у детей с отклонениями в поведении.
Исследование когнитивных вызванных потенциалов головного мозга входит в обязательную программу обследования детей с ДЦП и ЗПР, поступающих на реабилитацию, что дает возможность оценить степень поражения высших корковых функций и увидеть положительные изменения после курса комплексного лечения.
Использование специалистами МЦ «CORTEX» различных методик оценки функционального состояния центральной нервной системы позволяют провести диагностику, составить наиболее оптимальный для каждого пациента лечебный комплекс и провести оценку его эффективности, а также разработать план дальнейшей реабилитации.
Электронейромиография
Электронейромиография (ЭНМГ) – это комплексное исследование, при помощи которого определяют общее функциональное состояние периферической нервной системы и мышц. В МЦ «CORTЕХ» исследование проводится с использованием многофункционального компьютерного комплекса для миографии и исследования вызванных потенциалов мозга — «Нейро –МВП».
Объединяет в себе два метода исследований, что дает более полную картину:
1.Электромиография (ЭМГ) – это аппаратный метод исследования биоэлектрической активности мышц, с помощью которого определяют потенциал двигательной единицы в состоянии покоя и во время сокращения. Как известно, каждая мышца содержит разное количество волокон, от 7 до 2.000, которое зависит от вида мышцы. Сокращаясь синхронно, мышечные волокна образуют потенциал двигательной единицы, который является суммой потенциалов мышечных волокон. Размеры и форма потенциалов может изменяться при различных заболеваниях периферической нервной системы. По этим изменениям можно судить о состоянии периферической и центральной нервной системы. Амплитуда колебаний мышечного потенциала составляет всего несколько милливольт, а длительность – не больше 25 мс. Электромиограф улавливает и визуализирует их на фотопленке в виде кривой — электромиограммы.
2.Электронейрография (ЭНГ) – это аппаратный метод, позволяющий измерить скорость проведения электрического импульса по нервам. Мышцы, как и другие органы-исполнители, связаны с центральной нервной системой с помощью периферических нервов. Сигнал передается по нервам к спинному и головному мозгу, то же самое происходит и в обратном направлении. Во время исследования проводят стимуляцию периферического нерва и замеряют уровень активности в двух других точках по пути ее следования.
Суть методики: Нервная система нашего организма состоит из двух частей, которые функционально связаны друг с другом – центральная нервная система и периферическая. Связь между ними осуществляется посредством электрических импульсов, передающихся по нервам от нервных окончаний к головному и спинному мозгу. Все наши ощущения — это информация, полученная от рецепторов и переданная в мозг. При патологиях или заболеваниях нарушаются пути следования импульса и способность правильного восприятия информации. При повреждении или нарушении пути следования импульса от головного или спинного мозга к мышцам человек либо вообще теряет способность двигаться, либо не может это делать полноценно. Проявлениями таких заболеваний могут быть паралич, слабость мышц, парезы.
Во время исследования стимулируется отдельный нерв и регистрируется ответ соответствующей мышцы, иннервируемой этим нервом. Например, при исследовании головного мозга стимулируют слуховые, зрительные нервы и анализируют ответ центральной нервной системы.
ЭНМГ является самым информативным методом исследования, помогающим диагностировать заболевания верхних и нижних конечностей, суставов и мышц.
Методика помогает выявить заболевание на ранней стадии, что существенно облегчает излечение пациента. Ни один метод исследования не может дать такой полной информации относительно состояния аксона. Электронейромиография помогает определить, в какой части нерва находится проблема и насколько она серьезна.
Также ЭНМГ позволяет наблюдать за изменениями состояния пациента во время лечения и эффективности тех или иных методов терапии.
ЭНМГ проводиться тремя способами:
Поверхностная. Импульсы передаются посредством накожных электродов, закрепленных на верхних и нижних конечностях. Это неинвазивный способ без стимуляции. Метод достаточно простой и широко применяется при медицинских экспертизах.
Игольчатая. Инвазивный метод с введением игольчатых электродов непосредственно в мышцу и определяющий ее активность.
Стимуляционная. Этот вид электронейромиографии отличается от поверхностной проведением стимуляции нервных волокон. Исследование проводят с помощью накожных и игольчатых электродов.
Показания для диагностики:
- радикулит– невралгическое заболевание, возникающее в результате повреждения или защемления корешков спинномозговых нервов (шейный, грудной, поясничный радикулит);
- туннельный синдром – защемление срединного нерва костями запястья и сухожилиями мышц;
- невропатии – врожденные или приобретенные дисфункции нервов, причинами которых могут быть инфекционные заболевания, травмы, сахарный диабет;
- боковой амиотрофический склероз (БАС) – неизлечимое поражение спинного и продолговатого мозга;
- плексопатии – поражение нервных сплетений в результате травмы, злокачественной опухоли, лучевой терапии, результатом которого может быть паралич.
Противопоказания к проведению диагностики:
Процедура противопоказана больным с эпилепсией и психическими расстройствами, а также гипертонией.
Никакой особенной подготовки для проведения исследования не требуется. Длительность сеанса – 30-60 минут в зависимости от вида ЭНМГ и размера обширности исследования. В время исследования пациент находится на кресле в состоянии лежа или полусидячем. Поверхностное исследование совершенно безболезненно. Небольшие болевые ощущения могут присутствовать в момент введения игольчатого электрода во время игольчатой или стимуляционной ЭНМГ. Доктор определяет точки крепления электродов. Эти места предварительно протираются дезинфицирующим раствором и смазываются специальным гелем. После исследования пациент может чувствовать небольшую слабость в мышцах.
Расшифровка результатов ЭНМГ проводится только врачом функциональной диагностики. Он сверяет полученные показания с нормой, определяет степень отклонения и на основании этих данных ставит диагноз.
Полуколичественное определение в крови аутоантител класса IgG, взаимодействующих с антигенами нейронов (белок NF200), глиальных клеток (GFAP), нервных волокон (ОБМ) и рецепторами нейромедиаторов.
«ЭЛИ-Тесты» позволяют на основании оценки состава крови заметить патологии еще на том этапе развития, когда отсутствуют внешние симптомы и человек чувствует себя здоровым. Данный тест позволяет оценить состояние центральной и периферической нервной системы по маркерам-антителам. Исследование особенно значимо, когда человек находится в группе риска (наследственная предрасположенность) к тяжелым патологиям нервной системы, таким заболеваниям как инсульт, боковой (латеральный) амиотрофический склероз. Определяется 12 видов аутоантител.
Синонимы русские
ЭЛИ-Н-Тест-12, ЭЛИ-НейроТест-12, состояние нервной системы, нейронный тест, нейротест, биомаркеры нервной системы.
Синонимы английские
Analysis of the nervous system, evaluation of 12 antigens.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
Подготовки не требуется.
Общая информация об исследовании
Определяемые аутоантитела:
|
Функционально-клинические характеристики антител (АТ) |
АТ к антигену |
|
АТ-маркеры дегенеративных процессов в аксонах |
NF200 |
|
АТ-маркеры глиоза |
GFAP |
|
АТ-маркеры нарушений эмоционального статуса, ассоциированные с ВПЧ-инфекцией |
S100 |
|
АТ-маркеры нарушений миелиновой оболочки аксонов |
ОБМ |
|
АТ-маркеры нарушений нейромышечных контактов |
(V)-Ca-канал |
|
АТ-маркеры миастенических синдромов и нарушений механизмов обучения и памяти |
Н-Холино-Рц |
|
АТ-маркеры нарушений процессов регуляции торможения/возбуждения в ЦНС |
Глутамат-Рц |
|
ГАМК-Рц |
|
|
АТ-маркеры нарушений мотивационно-волевой сферы |
Дофамин-Рц |
|
АТ-маркеры нарушений эмоциональной сферы |
Серотонин-Рц |
|
АТ-маркеры нарушений эмоционально-мотивационной сферы |
Опиантные Рц |
|
B-Эндорфин |
Естественные аутоантитела (ауто-АТ) класса IgG разной антигенной (органной, тканевой) специфичности постоянно синтезируются в организме любого здорового человека и участвуют в клиренсе организма от продуктов обмена, а также в регуляции функций клеток разных типов. Развитие любой болезни сопровождается патологической активацией гибели (апоптоз, некроз) клеток определенных органов и увеличением выброса соответствующих антигенов и/или изменением синтеза и секреции определенных макромолекул. Это влечет за собой вторичные изменения продукции ауто-АТ соответствующей специфичности. Избирательное повышение или снижение иммунореактивности (сывороточного уровня) отдельных ауто-АТ может указывать на изменения, затрагивающие определенные органы и ткани. Стойкие изменения в содержании ауто-АТ могут предшествовать клинической манифестации патологии (в некоторых случаях за месяцы и годы до развития заболевания), что позволяет использовать их в прогностических целях. Для одновременной оценки содержания множества ауто-АТ используются методы группы «ЭЛИ-Тест». Они не заменяют другие методы обследования, но позволяют подойти к их назначению наиболее обоснованно и адресно, с учетом индивидуальных показаний.
Серьезные заболевания могут развиваться постепенно и в начале могут быть незаметными самому человеку и при врачебных обследованиях со стандартной диагностикой. Однако в это время клетки того органа или систем, которые будут затронуты болезнью, уже начинают изменять состав продуктов обмена, выделяемых в кровяное русло и затем появляются определенные аутоантитела для утилизации этих секретируемых продуктов. По определению в крови таких биомаркеров можно выявить уже существующую угрозу того или иного заболевания.
Функциональную деятельность организма осуществляет нервная система, которая объединяет центральный (спинной и головной мозг) и периферический (нервные волокна, поддерживающие двигательную активность тела) отделы. Для раннего диагностирования патологических процессов, которые уже без видимых проявлений развиваются в нервной системе, используют «ЭЛИ-Н-ТЕСТ». Он позволяет оценить по маркерам-антителам состояние центральной и периферической нервной системы. Исследование особенно значимо, когда человек находится в группе риска (наследственная предрасположенность) к таким заболеваниям как инсульт, боковой (латеральный) амиотрофический склероз. Определяется 12 видов аутоантител.
Для чего используется исследование?
Позволяет диагностировать на ранних этапах:
- дегенерацию нервных волокон;
- аномально активное разрастание клеток астроглии (глиоз);
- патологический процесс в нервных волокнах, например при демиелинизации;
- риск развития бокового (амиотрофического)склероза, синдрома Ламберта – Итона или мозжечковой атаксии;
- неспецифические инфекции и вызванное ими воспаление.
Когда назначается исследование?
- Первичный скрининг состояния организма (наиболее обоснованное и адресное назначение других методов обследования, с учетом индивидуальных показаний).
- Уточнение диагноза в сложных случаях.
- Целесообразность начала применения фармакологического лечения, а также его назначения в минимально необходимой дозировке.
- Опережающий мониторинг изменений состояния больного под влиянием лечения, т.е. объективная оценка эффективности назначенного лечения и его достаточности.
Что означают результаты?
|
Значение |
Интервал (для АТ-маркеров) |
|
Норма |
от — 15 % до 10 % |
Интерпретация результатов
|
NF-200 |
Специфический белок аксонов. |
|
GFAP |
Специфический белок филаментов астроцитов. |
|
S100 |
Белок S100 – регулятор дифференцировки и морфогенеза многих типов клеток (в том числе нервных клеток плода). |
|
ОБМ |
Общий белок миелина — специфический антиген миелиновых оболочек аксонов. Процессы патологических |
|
(V)-Ca-канал |
Вольтажзависимый Са-канал. |
|
н-Холино-Рц |
н-Холино-Рц (никотиновые ацетилхолиновые рецепторы); глутамат-Рц (глутаматные рецепторы); ГАМК-Рц (ГАМК-рецепторы); дофамин-Рц (дофаминовые рецепторы); серотонин-Рц (серотониновые рецепторы). |
|
Глутамат-Рц |
|
|
ГАМК-Рц |
|
|
Дофамин-Рц |
|
|
Серотонин-Рц |
|
|
Опиатные-Рц |
Опиатные-Рц (опиатные рецепторы) и B-Эндорфин. |
|
B-Эндорфин |
Повышенные уровни специфических ауто-АТ
Столбики гистограммы, обращенные в положительную область от уровня индивидуальной средней иммунореактивности (обозначен нулевым уровнем на гистограмме).
- При нормальном состоянии органов и систем отмечаются лишь небольшие динамические колебания сывороточных концентраций органоспецифических ауто-АТ в пределах «зеленой зоны» вокруг индивидуальной средней.
- Важную прогностическую и клиническую значимость имеют столбики гистограммы, выходящие в зону за пределами оптимальных значений (+10 %) и особенно превышающие значение + 15 %. Умеренное повышение ауто-АТ определенной органной специфичности, (градиентный цветовой переход на графике от зеленого к красному) говорит о наличии в затронутом органе патологического процесса относительно небольшой интенсивности, который может развиться в клинически выраженную симптоматику при длительном сохранении изменений.
- Высокий титр ауто-АТ соответствующей специфичности (столбики достигают «красной зоны») говорит об активном патологическом процессе повышенной интенсивности.
Пониженые уровни специфических ауто-АТ
Столбики гистограммы, направленные в отрицательную область от уровня индивидуальной средней иммунореактивности. Уровни ауто-АТ, выходящие в зону за пределами оптимальных
Значений (-15 %) и особенно опускающиеся ниже — 20 %, сопровождают развитие патологии и обычно указывают на:
- избыточный выброс антигена (например, при интенсивном распаде ткани рак, туберкулез и т.д.);
- избыток антиидиотипических антител (при длительном, хроническом, постепенно затухающим патологическом процессе);
- нефизиологическое снижение (индивидуальные особенности реагирования иммунной системы) синтеза и секреции ауто-АТ.
Изменения гистограммы во времени (динамика)
Динамические изменения отражают интенсивность и выраженность патологического процесса, включая изменения объема очага поражения.
Отсутствие изменений на гистограмме при подтвержденной патологии органа
Указывает на транзиторное или постоянное прекращение патологического процесса к моменту исследования.
Также рекомендуется
- Антитела к кардиолипину, IgG и IgM
- Антитела к β2-гликопротеину I, IgG и IgM
- Иммуноблот антифосфолипидных антител
- Иммуноблот антинуклеарных антител
- Фенотипирование лимфоцитов (основные субпопуляции) — CD3, CD4, CD8, CD19, CD56
Кто назначает исследование?
Невролог, психиатр.
Литература
- A Manual of Laboratory and Diagnostic Tests, 9th Edition, by Frances Fischbach, Marshall B. Dunning III. Wolters Kluwer Health, 2015. Page 606-607.
- Henry’s Clinical Diagnosis and Management by Laboratory Methods, 23e by Richard A. McPherson MD MSc (Author), Matthew R. Pincus MD PhD (Author). St. Louis, Missouri : Elsevier, 2016. Pages 995, 1003-1004.
- Клиническая лабораторная диагностика: национальное руководство: в 2 т. – T. II / Под ред. В. В. Долгова, В. В. Меньшикова. – М.: ГЭОТАР-Медиа, 2012. С. 82-84, 99.
- Инструкция по применению набора реагентов для полуколичественного определения в сыворотке крови обследуемых пациентов панели аутоантител-маркеров состояния иммунной системы «ЭЛИ-Вакцина-ТЕСТ».
Исследование проводимости периферических нервов и электромиография
Исследование проводимости периферических нервов позволяет просто и надежно определить состояние периферических нервов. Импульс, вызванный электростимуляцией нерва, направляется по двигательным, чувствительным и смешанным нервам, и характеристики проведения импульса оцениваются с помощью записи потенциалов с мышц, либо непосредственно с нерва.
Двигательная единица состоит из одиночного нижнего двигательного нейрона и всех иннервируемых им мышечных волокон. Исследование проводимости двигательного нерва используется для оценки целостности двигательной единицы. При этом исследователь получает информацию о функционировании и структурной целостности двигательного нейрона, нерва, нервно-мышечного соединения и мышцы. Она позволяет установить локализацию, распространенность, длительность и патофизиологические особенности повреждений периферической нервной системы (ПНС). Также можно получить представления о прогнозе, эффективности лечения и степени восстановления двигательной единицы. При исследованиях двигательной проводимости записывающие электроды размещают на коже над мышцей и сухожилием, а стимулирующие электроды размещают на коже вдоль исследуемого нерва. Ответ мышцы на электростимуляцию может быть измерен путем регистрации суммарного потенциала действия мышцы (СПДМ), являющегося суммой электрических потенциалов всех мышечных волокон, которые реагируют на стимуляцию нерва. Может быть определено время, необходимое электрическому импульсу для достижения мышцы (латентность). Скорость прохождения импульса по нерву определяют путем стимуляции нерва в различных местах и определения дистанции, которую стимул преодолел.
Исследование проводимости двигательного нерва могут быть использованы в следующих целях:
- Для получения объективных доказательств поражения двигательной единицы.
- Для идентификации и определения точного места компрессии, ишемии и очаговых повреждений нервов, которые могут проявляться блокадой проведения импульса, замедлением проведения импульса в месте повреждения или патологическим проведением проксимальнее или дистальнее повреждения.
- Для определения степени распространенности поражения нервов у пациентов, у которых наблюдаются признаки поражения одиночного нерва (например, при мононевропатиях).
- Для дифференциальной диагностики периферических невропатий, миопатий и болезней нижнего двигательного нейрона (например, бокового амиотрофического склероза) у пациентов со слабостью конечностей.
- Для диагностики заболевания до его перехода в стадию развернутых клинических проявлений (например, при семейных невропатиях).
Обследование по поводу заболеваний нервно-мышечного синапса может включать ритмическую стимуляцию двигательных нервов. По мере утомления нервно-мышечного соединения при записи СПДМ, и его сравнении с полученным позднее СПДМ может наблюдаться падение амплитуды потенциала, поскольку со временем все меньше и меньше волокон способны реагировать на стимуляцию, даже если стимулировать нерв с интенсивностью, которую в норме нерв способен выдерживать длительное время.
Исследования проводимости чувствительных нервов проводятся с помощью записи потенциалов действия, при электростимуляции кожного нерва. Селективные исследования чувствительных нервов могут быть выполнены при стимуляции нервов, имеющих только чувствительный компонент (например, икроножного нерва), или, в качестве альтернативы, при селективной стимуляции чувствительного компонента смешанного нерва. Последнее, может быть сделано путем анатомической изоляции чувствительного компонента (например, стимуляция пальцев руки и запись над смешанным нервом в области запястья или локтя) или стимуляции смешанного нерва и записи над пальцами, в области которых расположены преимущественно чувствительные аксоны.
Исследования проводимости чувствительных нервов могут представлять ценность в следующих случаях:
- При системных заболеваниях, протекающих с поражением чувствительных нервов— для определения разновидности вовлеченных в патологический процесс чувствительных нервов (например, тонкие волокна, проводящие болевые и температурные ощущения, или толстые волокна, ответственные за проприоцептивную чувствительность); для установления того, какая часть периферического нерва поражена в большей степени — аксон или миелиновая оболочка; для получения объективных доказательств поражения чувствительного нерва.
- При очаговых невропатиях — для определения места повреждения или блокады, особенно при изолированном поражении чувствительных нервов.
- Для подтверждения или количественной оценки нарушений, если расстройства чувствительности возникают при периферической невропатии раньше, чем двигательные изменения или до появления объективных клинических признаков.
- Для установления локализации повреждения (проксимальнее или дистальнее) по отношению к ганглию заднего корешка (например, при дифференциальной диагностике повреждения плечевого сплетения и повреждения корешков).
Электромиографию (ЭМГ) обычно выполняют вместе с исследованиями проводимости нервов, получая при этом дополнительную информацию. Игольчатый электрод вводят в исследуемую мышцу и регистрируют потенциалы действия, генерируемые группами мышечных волокон (потенциалы действия двигательной единицы, или ПДДЕ). Исследуют мышцы в покое, в состоянии слабого сокращения и в состоянии сильного сокращения. В норме в состоянии покоя активность мышц не регистрируется. При активно протекающей невропатии, при тяжелых или воспалительных миопатиях могут регистрироваться спонтанные потенциалы действия с одиночных мышечных волокон (фибрилляционные потенциалы). При некоторых неврогенных процессах (особенно это характерно для болезни двигательного нейрона) могут наблюдаться спонтанные сокращения групп мышечных волокон (фасцикуляционные потенциалы). Характерные изменения ПДДЕ могут наблюдаться при патологии нервов и мышц. При заболевании периферических нервов амплитуда, продолжительность и степень полифазности ПДДЕ часто увеличены, а восстановление затруднено, в то время как при миопатиях амплитуда и продолжительность ПДЕ могут быть снижены, полифазность увеличена, восстановление ускорено. Потенциалы действия единичного мышечного волокна могут быть исследованы с помощью технически более сложного метода — электромиографии одиночного мышечного волокна.
В целом, электромиография и исследования нервной проводимости используются для обследования и уточнения диагноза у пациентов с болезнью двигательного нейрона (например, при боковом амиотрофическом склерозе), патологическими процессами, протекающими с поражением сплетений или нервных корешков, компрессионными невропатиями, периферическими полиневропатиями, заболеваниями нервно-мышечного синапса (например, myasthenia gravis), а также с заболеваниями мышц. Поскольку исследование требует введения игольчатых электродов в мышцы и применения электрических разрядов, для пациента оно сопряжено с определенными неудобствами. При соблюдении техники безопасности исследование не представляет опасности; ограничить проведение ЭМГ может склонность пациента к кровотечениям.
ЭМГ и определение скорости распространения возбуждения (СРВ) по нервному волокну при различных заболеваниях
1. ЭМГ и исследование СРВ важны при обследовании и электрофизиологической диагностике болезней двигательного нейрона (например, бокового амиотрофического склероза). В целом, исследования проводимости периферических нервов дают нормальные результаты, кроме, вероятно, некоторого снижения амплитуд ПДЕ (поскольку заболевание исключительно двигательного характера, результаты исследования чувствительности патологии не выявляют). С помощью игольчатой ЭМГ можно обнаружить признаки диффузного повреждения клеток переднего рога, в том числе патологическую спонтанную активность (фибрилляции и фасцикуляции), патологические параметры (увеличение амплитуды, расширение, полифазность) и замедление восстановления ПДЕ. Часто данные ЭМГ свидетельствуют об активном патологическом процессе даже при отсутствии клинических проявлений заболевания или минимальных проявлениях. С помощью игольчатой ЭМГ можно получить также информацию о прогнозе заболевания; ЭМГ может помочь диагностировать другие заболевания клеток переднего рога, такие как постполиомиелитический синдром и спинальная мышечная атрофия.
2. Термин радикулопатии объединяет различные симптомы и признаки, возникающие в результате преходящего или стойкого повреждения нерва при его выходе из спинного мозга на уровне межпозвоночных отверстий. Результаты исследований проводимости обычно в норме. ЭМГ выявляет признаки неврогенных изменений (например, фибрилляции и изменения ПДЕ) в мышцах, иннервируемых определенным корешком, тогда как мышцы, иннервируемые не вовлеченными в патологический процесс корешками, интактны. Характер неврологических изменений зависит от степени тяжести процесса, длительности заболевания и степени восстановления (реиннервации).
В клинической практике ЭМГ может быть полезна в следующих ситуациях:
- ЭМГ используется для подтверждения повреждения корешка и определения уровня поражения. Следует заметить, что патологические изменения по результатам ЭМГ наблюдались лишь примерно у 90 % пациентов с шейной или пояснично-крестцовой радикулопатиями, обнаруженных при оперативном вмешательстве. Таким образом, нормальные результаты ЭМГ не исключают наличия радикулопатии.
- ЭМГ позволяет уточнить вовлечение конкретных корешков.
- ЭМГ используют для выявления активной денервации (определяется по наличию фибриллярных потенциалов).
- С помощью ЭМГ можно определить время, прошедшее с момента возникновения радикулопатии (острая, подострая, хроническая или длительно существующая).
- ЭМГ может предоставить определенную информацию о степени выраженности радикулопатии.
- При ЭМГ можно обнаружить другую патологию, способную объяснить существование имеющихся у пациентов симптомов.
- ЭМГ может помочь определить, имеют ли обнаруженные при МРТ или миелографии изменения какое-либо физиологическое значение.
- Помощью ЭМГ и ЭНМГ можно диагностировать плечевые и пояснично-кресцовые плексопатии и компрессионные нейропатии, а также определить уровень поражения при этих заболеваниях.
3. ЭМГ и ЭНМГ часто назначают при периферических полинейропатиях. Электрофизиологические характеристики невропатий используют в качестве дополнительной информации для уточнения природы заболевания, что позволяет сузить круг дифференциально-диагностического поиска. Результаты ЭМГ/ ЭНМГ позволяют оценить степень заинтересованности двигательных и чувствительных нервов; определить, является ли поражение главным образом результатом повреждения миелиновой оболочки или аксона; указать, является повреждение очаговым или диффузным; определить, распространяется процесс дистальнее или проксимальное; предоставить информацию о степени тяжести и длительности существования патологического процесса. Может наблюдаться увеличение дистальных чувствительных и двигательных латентностей, замедление скорости проведения, патология чувствительных ответов и ПДДЕ и «неврогенные» ЭМГ — изменения. Патологические результаты исследований подтверждают наличие невропатий, но следует заметить, что при невропатиях с поражением тонких чувствительных волокон (проводящих болевую и температурную чувствительность) результаты исследований часто нормальны. С помощью ЭМГ/ ЭНМГ можно дифференцировать генерализованную сенсомоторную периферическую полинейропатию от множественных мононейропатий в местах частой компрессии (например, невропатий срединного и локтевого нервов в области запястья).
По электрофизиологическим характеристикам периферические полинейропатии могут быть разделены на следующие категории:
- Демиелинизирующие смешанные сенсомоторные невропатии, в том числе некоторые наследственные невропатии,
- Сегментарные демиелинизирующие сенсомоторные полинейропатии, в том числе воспалительные невропатии (например, синдром Гиен-Барре) и невропатии, ассоциированные с гаммапатиями, гипотиреозом, злокачественной опухолью или лимфомой, СПИДом, болезнью Лайма и воздействием определенных токсинов.
- Аксональные моторно-сенсорные полинейропатии, включая порфирию, некоторые наследственные невропатии, лимфоматозные невропатии и некоторые токсические невропатии.
- Аксональные сенсорные нейронопатии или нейропатии, включая первичный амилоидоз, синдром Шегрена, паранеопластические нейропатии, а также нейропатии, вызванные приемом лекарственных препаратов и дефицитом витамина В12.
- Смешанные аксональные сенсомоторные полинейропатии, в том числе нейропатии при уремии и сахарном диабете.
- Аксональные сенсомоторные полинейропатии, в том числе нейропатии, вызванные дефицитом определенных питательных веществ, приемом алкоголя, связанные с саркоидозом, заболеваниями соединительной ткани, воздействием токсинов, тяжелых металлов и лекарственных препаратов.
4. Заболевания нервно-мышечного синапса могут быть диагностированы с помощью ритмической стимуляции. Ритмическая стимуляция двигательных нервов применяется, в основном, для диагностики миастении. Для этой патологии характерно прогрессивное снижение амплитуды ответа на несколько первых раздражающих стимулов, получаемое при стимуляции с частотой 3 стимула в секунду. Уточнить характер заболевания можно по изменению ответа на стимуляцию после непродолжительного сокращения мышцы. У некоторых пациентов с миастенией при нормальных результатах стимуляции диагноз может быть установлен с помощью ЭМГ единичного мышечного волокна. При миастеническом синдроме Итона-Ламберта значительно уменьшена амплитуда ответа находящейся в покое мышцы, вызванного единичной максимальной стимуляцией нерва. Дальнейшее уменьшение амплитуды может наблюдаться при ритмической низкочастотной стимуляции, но значительное улучшение (увеличение ПДДЕ) наблюдается во время высокочастотной стимуляции. При других заболеваниях, таких как боковой амиотрофический склероз, иногда может наблюдаться необычная утомляемость периферической нервно-мышечной системы, но это патологическое изменение не представляет большой диагностической ценности.
5. У пациентов с миопатиями, электродиагностические исследования демонстрируют широкий спектр отклонений. Основные параметры ЭНМГ в норме, за исключением иногда наблюдающегося снижения амплитуды моторных ответов. При ЭМГ могут регистрироваться фибриллярные потенциалы при тяжелых миопатиях или воспалительных миопатиях (например, полимиозите). «Миопатический» ПДЕ характеризуется снижением амплитуды и продолжи¬тельности с увеличением полифазии и быстрым восстановлением вне зависимости от степе¬ни сокращения мышцы. Одной ЭМГ обычно недостаточно для диагностики заболевания, но результаты ЭМГ могут быть использованы для отнесения патологии к определенной группе мышечных нарушений. Токсические и эндокринные миопатии могут не сопровождаться патологическими отклонениями на ЭМГ, или эти отклонения оказываются весьма незначительными.
ЭМГ/ЭНМГ позволяют прояснить следующие вопросы
- Дифференцировать нейрогенные и миопатические причины мышечной слабости.
- Установить ключевые моменты этиологии миопатии.
- Оценить тяжесть патологического процесса, а также является ли он острым, подострым или хроническим.
- Оценить степень активности и характер течения заболевания.
- Предоставить информацию о распространенности патологического процесса.
- Диагностировать патологию, даже если она не сопровождается клиническими проявлениями.
Автор: врач-невролог высшей квалификационной категории Трубицына О.В.
Тел.: 267-36-66, 267-09-11
Lindsey R. FISCHER, Jonathan D. GLASS, Departments of Neurology, and Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, USA
Резюме
Данные, полученные при исследовании бокового амиотрофического склероза у животных и людей, все больше подтверждают предположение о том, что дистальная дегенерация аксонов при этом заболевании начинается на исключительно ранних стадиях. Это происходит гораздо раньше появления первых признаков гибели двигательных нейронов и возникновения отчетливой симптоматики. Причина аксональной дегенерации в настоящее время не выяснена. Возможно, что сочетание нескольких процессов приводит к развитию данного состояния. Вовлечение местных повреждающих факторов и резкое уменьшение трофической поддержки по причине изменения метаболизма окружающих тканей являются обязательными компонентами при развитии данной патологии. Совершенно понятно, что аксон по своей природе не является простым придатком тела нейрона, его дегенерация и гибель подчиняются тем же законам и правилам. Эти данные подтверждаются рядом исследований, в которых использовались объекты с искусственно созданным состоянием амиотрофического латерального склероза. При анализе указанных данных, связанных с методами защиты двигательных нейронов от любых травмирующих факторов, было установлено, что они не привели к сколько-нибудь значимым изменениям симптоматики или удлинению времени нормального функционирования исследуемых нейронов. Аналогичные данные были получены при попытке использовать технологии по защите непосредственно аксона, исследуемой клетки. В настоящей статье приводится развернутый анализ состояний ранней аксональной дегенерации при боковом амиотрофическом склерозе и обсуждаются возможные механизмы развития данного состояния с акцентированием внимания на процессы оксидативного стресса. Обсуждается первичное значение аксональной дегенерации как возможного механизма развития данного заболевания двигательных нейронов. Кроме того, предлагаются к рассмотрению факторы, предупреждающие развитие аксональной дегенерации, как важный аспект терапии данного состояния.
Ключевые слова
боковой амиотрофический склероз, аксональная дегенерация, нейромышечная связь, супероксиддисмутаза, оксидантный стресс
Введение
Аксональная дегенерация является одним из основных клинических и патологических факторов развития бокового амиотрофического склероза (БАС). Аксональная дегенерация в основном заканчивается дисфункцией клетки или ее гибелью, связанной с валлеровской или подобной дегенерацией. Сейчас опубликовано множество работ, касающихся экспериментальных данных, которые говорят, что любая защита тел мотонейронов от внешних воздействий имеет исключительно малую эффективность по отношению к скорости развития симптомов БАС. Фактически даже генетические вмешательства, которые, казалось, могли бы полностью изменить скорость развития патологического состояния в мотонейронах спинного мозга, до сих пор не в состоянии предотвратить или замедлить ослабление и гибель животных в экспериментальной модели заболевания мотонейронов. Этот кажущийся парадокс может быть объяснен тем, что невозможно уберечь тела нейронов от патологических процессов, которые уже развились в их аксонах. Прямым следствием повреждения аксонов является дегенеративный процесс, развивающийся в мышечной ткани. Указанные факты еще раз подчеркивают выдвинутую не так давно идею о том, что дегенерация аксонов двигательных нейронов не является прямым следствием дегенерации и гибели тела родительского нейрона. Кроме того, подчеркивается, что даже незначительных изменений в аксональном метаболизме будет достаточно для формирования клинической картины. Именно поэтому предупреждение аксональной дегенерации является важным с терапевтической точки зрения. Успех в достижении этой цели даст возможность улучшить терапию БАС и других нейродегенеративных заболеваний.
Аксональная дегенерация при раннем БАС
Что общего между аксональной дегенерацией двигательных нейронов при моделировании БАС и аналогичными состояниями у человека?
Моделирование на животных
Развитие патологического процесса, сопровождающегося нейрональной дегенерацией, в настоящее время широко исследуется на животных при моделировании данной патологии. Важную роль занимают также исследования, проводимые на пациентах с нейродистрофической патологией и с невропатией двигательных нейронов спинного мозга в частности. В подавляющем большинстве случаев развитие нейродистрофии является первичным процессом, обусловливающим недостаточность функции нейронов и проявление симптомов заболевания. При этом даже весьма выраженные парезы сопровождаются неожиданно легкими изменениями двигательных нейронов. Данные явления некоторые авторы объясняют изменениями в генетическом аппарате клеток, обусловливающими выраженные изменения их функций с легкими морфологическими отклонениями. Аксональная дегенерация, несомненно, является следствием аналогичных патологических процессов, приводящих к гибели нейрона, что, в свою очередь, вызывает денервацию соответствующей мышцы. Данные, касающиеся экспериментальных моделей, на которых производился анализ описанных ситуаций, приведены ниже.
Для анализа наследственного БАС наиболее широко используется моделирование на мутантных линиях мышей по Cu,Zn-супероксиддисмутазе. Высокая степень копирования мутантной SOD1 вызывает появление слабости на 80–90-й день и гибель животного на 130–140-й день. Оригинальные исследования показывают, что начало развития клинической картины у таких животных совпадает по времени с началом частичной гибели мотонейронов спинного мозга. При этом сопутствующие физиологические и патофизиологические исследования выявляют начало денервации мышц задних конечностей задолго до появления характерных симптомов заболевания. Для установления пространственно-временных особенностей развития процессов, приводящих к гибели мотонейрона, было проведено количественное исследование патологических процессов от конечной до начальной точки их развития. Значительные явления мышечной дегенерации появились на 47-й день, задолго до появления первых симптомов заболевания. На момент появления первых клинических признаков (80-й день исследования) две трети аксонов вентральных корешков уже были подвергнуты дегенерации. До момента развития ярких симптомов заболевания не встречалось ни одного случая выявления гибели тел мотонейронов. Аналогичные данные были представлены и другими лабораториями, которые работают с мутантными линиями мышей различных подтипов.
В серии работ использовались животные со специфической миссенс-мутацией, сопровождающейся дефектом синтеза тубулина в нейронах, что приводило к прогрессирующей моторной невропатии. У таких животных развитие слабости происходило в течение первых трех недель жизни, и гибель животных происходила на шестой неделе. Гибель мотонейронов или даже развитие дегенерации дистальной части аксонов не отмечались на фазе развития явных патологических проявлений. В конечной стадии развития данной патологии гибель мотонейронов встречалась не более чем в 40 % всех случаев. Электрофизиологические исследования показали некоторые изменения структуры потенциала действия в моторных нейронах, иннервирующих мускулатуру головы и задних конечностей животного. При этом прослеживались временные зависимости нарастания указанных изменений. Причем самые выраженные изменения наблюдались задолго до тринадцатого дня исследования. Очевидные патологические изменения, свидетельствующие о денервации указанных мышц, наблюдались после пятнадцатого дня исследования. Таким образом, можно сделать вывод об исключительно малой корреляции между выраженностью патологического процесса и клиническими проявлениями у данных животных.
Группа животных с моделированной спинальной мышечной атрофией, вызванной искусственным удалением седьмого экзона гена Smn, характеризовалась ранней развивающейся моторной деградацией и гибелью животного в возрасте около четырех недель. Патоморфологический анализ данных животных выявил, что было потеряно только около 30 % поясничных двигательных нейронов за тридцатидневный период. Кроме того, 49 % аксонов передних рогов сегментов L4–L5 подверглись деструкции в первые пятнадцать дней. Данный показатель вырос до 78 % к тридцатому дню. Денервация закономерно сопровождалась выраженным накоплением фосфорилированных нейрофиламентов в нейромышечных синапсах. Во всех случаях не было обнаружено явлений терминального прорастания, что может быть связано с сопровождающимся дефектом аксонального роста и пластичности. Анализируя спинальный уровень у данных животных, можно с уверенностью заключить, что дистальные аксональные дефекты были гораздо более выраженными по сравнению с деструкцией, происходящей в спинном мозге.
Во всех моделях большая часть двигательных нейронов была морфологически интактна в момент появления первых клинических признаков. Именно этот факт дает возможность предложить потенциально эффективное направление по предотвращению ухудшения состояния. При этом использование данных принципов у человека с БАС может помочь сохранить тела двигательных нейронов и при дальнейшей реиннервации улучшить состояние пациента.
БАС у людей
Аксональная дегенерация длительное время считалась единственной особенностью БАС у людей. При этом электромиографические исследования были основными в диагностике данных состояний. Такие особенности миограммы, как спонтанная фибрилляция и фастикуляция потенциалов, были наиболее важными в диагностике. Реиннервация при данном заболевании направлена на поддержание нормальной функции мышечной системы. Такое явление закономерно при БАС. Однако реиннервация не в состоянии адекватно компенсировать начавшуюся денервацию, которая и вызывает внезапную слабость соответствующих мышц. Денервацию традиционно приписывали формирующейся дисфункции и гибели мотонейронов. Помимо этого, реиннервация считалась следствием прорастания аксона моторного нейрона, лежащего в непосредственной близости от погибшего. Данные предположения не могли быть проверены на людях до момента смерти пациента с БАС.
Эти предположения поддерживаются еще и гипотезами о том, что БАС у людей начинается с дистальной дегенерации аксонов двигательных нейронов. Bradley с соавт. использовали количественные методики морфометрии для демонстрации проксимально-дистального градиента аксональной патологии в периферических нервах у людей с БАС. В нашем случае имелась редкая возможность оценить клинические, а затем патоморфологические данные у пациента с БАС, который умер во время оперативных вмешательств, не имеющих отношения к основному заболеванию. Клинический диагноз был установлен и прослежена клиника в течение не менее полугода с момента появления первых симптомов. Аутопсия подтвердила значительную денервацию и изменения в процессах реиннервации, которые были показаны миографически. Однако в данном случае не удалось выявить признаков, свидетельствующих об изменениях в мотонейронах спинного мозга.
В клинической практике исследователи использовали технологию определения числа моторных единиц для выявления количественных параметров процессов денервации и реиннервации. Данная технология позволила предположить, что уровень прогрессирования БАС у людей прямо зависит от денервации и потери функционирующих моторных единиц у пациента. Эти данные были проверены на линии мутантных мышей по супероксиддисмутазе-1. Технология подсчета моторных единиц использовалась и в этом случае. в ходе исследования получена важнейшая информация об уменьшении числа моторных единиц за несколько месяцев до начала формирования клинической картины. Для расширения спектра исследования использовалась методика порогового трекинга, которая позволяет выявить изменения аксональной возбудимости, что дало возможность получить также незаменимую информацию, касающуюся особенностей электрофизиологических изменений у пациентов с БАС. Явления резкого повышения натриевого тока и уменьшения калиевой проводимости наблюдались в двух популяциях пациентов. Особенностью этого состояния было значительное его усугубление в дистальных отделах аксонов. Несомненно, что ни одно из этих исследований не позволяет полностью раскрыть особенности течения БАС у людей. Однако в сочетании с данными, полученными на животных, можно с уверенностью говорить о том, что такое состояние является патологией дистальных отделов аксонов.
Последние исследования дали информацию еще об одном факторе, который может играть важную роль в формировании БАС. Именно информация о генетических изменениях при данной патологии заставляет все больше обращать на нее внимание. Говоря о генетических факторах, клиницисты должны иначе посмотреть на происхождение БАС. Из известных 43 мутаций, которые приводят к изменению двигательных нейронов, 14 причастны к формированию амиотрофического латерального склероза. Все мутации, имеющие место при БАС, влияют на такие принципиально важные моменты нейрональной биологии, как изменение цитоскелета нейронов, повреждение и трансформация медиаторных везикул, критические изменения аксонального транспорта. К роме того, важную роль в формировании патологического процесса играют нарушения ферментативных систем. Некоторые другие мутации вовлекают не только нейроны. В таких случаях причиной аномальной работы нервной клетки является дефект шванновских клеток. Роль этого типа глии сложно переоценить. Непосредственное участие в нейрональной передаче информации, в частности по моторным нейронам, осуществляется именно с помощью этих клеток. Все многообразие нарушений обмена клеток спинного мозга и окружающих тканей обусловливает постепенное развитие аксональной дегенерации, приводящей к моторной невропатии. Несмотря на все то обилие информации о явлениях, сопутствующих БАС, мы не можем сейчас более или менее четко сформировать стройную схему развития данной патологии. В наших силах проводить дополнительные исследования и подвергать более глубокому анализу уже полученные материалы.
Вовлечение сенсорных аксонов
Аксональная патология не ограничивается формированием БАС в эфферентном отделе ЦНС. Изменения в аксонах вовлекают и чувствительную часть нервной системы. Симптомы нарушения афферентных нейронов менее заметны и приводят к не настолько выраженным состояниям. Однако те изменения, которые встречаются у пациентов с БАС, также требуют коррекции. По усредненным данным, пациенты с БАС имеют снижение количества миелинизированных волокон в крупных нервных стволах до 70 % от исходного уровня. При этом количество данных волокон дорсальных корешков сегмента L5 уменьшается на 27 %. Изменения в телах нейронов, находящихся в околопозвоночных ганглиях на уровне 3–5-го поясничных сегментов, приводят к 54 % уменьшению их размеров. При анализе скорости проведения по сенсорным волокнам у пациентов с БАС выявляется значительное ее уменьшение в сравнении с контрольными группами пациентов. Исследования на мутантных животных выявили характерную особенность, которая заключалась в практически одновременном снижении количества моторных и сенсорных волокон при формировании БАС.
При анализе спинальной мышечной атрофии, вызванной прогрессированием БАС, выявлено, что имеет место снижение скорости проведения по смешанным нервам. При этом степень снижения колебалась от умеренной до выраженной. Одновременно проведенная биопсия выявила выраженное истончение нервных волокон. Гистологический анализ нервно-мышечных соединений у мутантных животных с БАС показал, что синаптические бляшки на соматической мускулатуре умеренно истончены, а данные, касающиеся анализа сенсорных волокон, свидетельствовали о практически полном исчезновении чувствительных нервных окончаний в верхних слоях кожи. В ходе анализа культуры нейронов, полученной от данных мышей, выявлялось, что их дендриты были значительно короче по сравнению с нормальными клетками. Аксональный холмик имел невыраженную структуру, количество т-РНК и м-РНК резко снижено в дистальных отделах аксонов. Обобщая полученные результаты, можно говорить об аналогичных процессах, происходящих в моторных и сенсорных нейронах, с учетом того, что в последних степень выраженности изменений несколько ниже.
Дегенерация сенсорных волокон при БАС не является прямым доказательством того, что именно двигательные нейроны являются местом развития всех патологических процессов при этом состоянии. Понимание причин того, что дегенеративные процессы развиваются в моторных аксонах при БАС с огромной скоростью и при этом сенсорные волокна остаются относительно интактными, дает ключ к дальнейшим изысканиям. Обнаружение причин и механизмов этих различий является первым шагом к лечению БАС. Больший размер двигательных нейронов, увеличенная метаболическая нагрузка, анатомическая организация, контакт с мышцами и другие отличия в структуре и функции двигательных нейронов рассматривались как причины формирования этой патологии. По нашим данным, прямые сравнения между чувствительностью моторных и сенсорных нейронов к действию таких потенциальных триггеров заболевания, как оксидативный стресс и экспрессия мутантной супероксиддисмутазы, проведены не были.
Механизмы аксональной дегенерации
При анализе литературы, касающейся влияния мутантной супероксиддисмутазы на дегенеративные процессы в аксонах, четкого описания этих процессов найдено не было. Кроме того, первичное место повреждения нервной системы также неизвестно. Отмирание части аксона может являться прямым следствием влияния травмирующего фактора на его дистальные отделы. Изучается и возможность перехода патологического процесса с тела нейрона на отростки. Системный или локальный принцип формирования БАС также рассматривался при анализе причин аксональной дегенерации. В данном разделе будут описаны механизмы дегенерации, присущие именно аксонам, а также будет рассмотрена возможность дистантного формирования БАС за счет использования мутантной супероксиддисмутазы как триггера аксональной дегенерации.
Аксональная дегенерация не зависит от гибели клеток
Очевидным является то, что аксональная дегенерация не является пассивным процессом. Она может быть лишь проявлением неизвестных нам процессов, часть из которых запрограммированна на гибель клетки. Например, удаление или уменьшение количества факторов роста нервной ткани из культуры клеток вызывает гибель тел клеток и их отростков. В опыте с мутантной линией мышей проводилось удаление проапоптотического гена, что вызвало сохранение двигательных нейронов у мышей с БАС. Однако денервация и дегенерация аксонов проявились вне зависимости от данной манипуляции. Все представленное выше подтверждает предположение о том, что аксональная дегенерация может происходить независимо от молекулярных процессов, контролирующих гибель клетки.
Концепция аксональной независимости была пересмотрена после открытия явления медленной валлеровской дегенерации, которая вызвана особым видом спонтанной мутации. Отличительной чертой в данном случае является удлиненная продолжительность жизни пораженных аксонов. Суть данного состояния сводится к тому, что мутантный ген обусловливает синтез нового химерного белка, имеющего протективное действие против химических и антигенных факторов, вызывающих аксональную дегенерацию. Несмотря на достаточно высокую эффективность данного белка, он все же не способен предотвратить дегенерацию или гибель клетки. Само существование такого агента, как химерный белок, имеющего уникальные свойства, свидетельствует о регулируемом процессе дегенерации клеток. Кроме этого, подтверждается гипотеза о различных механизмах клеточной и аксональной гибели. Эти данные позволяют предположить, что именно поиск в указанном направлении поможет найти инструмент для контроля за дегенеративными процессами в нервной ткани.
Эксперименты, проводящиеся на мутантных мышах с изменением в генотипе, подразумевающем наличие валлеровской дегенерации и прогрессирующей моторной невропатии, дали неоценимо важную информацию о разнонаправленности процессов сохранения тел и отростков нейронов. Мыши контрольной группы показали раннее развитие аксональной дегенерации в моторных нервах на сороковой день жизни. Введение дополнительной мутации, связанной с валлеровской дегенерацией, дало очень важный результат — неожиданно выраженное сохранение двигательных нейронов не только до сорокового дня, но и исключительно медленное развитие дегенеративных изменений, продолжавшихся до двух месяцев. Кроме этого, получены данные о том, что именно состояние аксонов прямо влияет на сохранность тела нейрона. Аналогичным образом проводилось тестирование групп мышей с мутацией по супероксиддисмутазе. Однако, к сожалению, при таком подходе было выявлено лишь незначительное изменение прогрессирования дегенерации, причем замедление этого процесса отмечалось только на первых этапах работы. Дополнительное исследование, проведенное с учетом всех поставленных задач, дало возможность достоверно предполагать, что протективные свойства атипичного белка, образующегося при валлеровской дегенерации, действенны первые две недели развития процесса. В последующие несколько месяцев сам химерный белок начинает работать как фактор, усугубляющий течение аксональной дегенерации. С учетом этих особенностей исследуемого белка был сделан вывод о возможности частичной приостановки подобного процесса дегенеративных изменений аксонов мотонейронов, который стремительно наблюдается при прогрессирующей моторной невропатии. К сожалению, использование этого же белка не имеет смысла в лечении хронической или вялотекущей дегенерации нервной ткани. Механизм действия протективного фактора, образующегося при валлеровской дегенерации, на сегодня не известен, однако его терапевтические эффекты могут найти применение в лечении огромного спектра патологических состояний. Определение гена или совокупности генов, ответственных за синтез протективного фактора при медленно прогрессирующей валлеровской дегенерации, не привело к успеху. Сейчас возможно его получение только при изменении достаточно крупного участка ДНК животного.
Роль кальпаина в развитии аксональной дегенерации
Исследования молекулярного механизма аксональной дегенерации поддерживают идею о неапоптотической программе гибели, развивающейся в аксоне. Практически при всех состояниях, ведущих к дегенеративным изменениям нервной клетки, первичными проявлениями являются замедление аксонального транспорта, митохондриальная дисфункция, увеличение концентрации кальция в клетке, что в конечном итоге приводит к активации системы кальпаинов. Кальпаины представляют собой группу нейтральных протеаз, активируемых кальцием. Их роль в формировании различных патологических процессов достаточно полно описана. Моделирование патологических состояний, проводившееся на животных и отдельных клетках, четко демонстрирует дегенерацию белков цитоскелета и мембраны клетки, вызванную кальпаином. При моделировании ишемических, травматических, токсических и других состояний результаты в отношении роли кальпаиновой системы были аналогичными. Ингибирование кальпаинов при периферической невропатии приводит к заметному снижению травмирующего влияния этих ферментов на клеточные элементы как в эксперименте на животном, так и на культуре клеток. Ингибирование кальпаинов также приводит к сохранению цитоскелета в нервно-мышечном соединении в модели острой аутоиммунной нервно-мышечной дегенерации. Полученные данные о роли кальпаинов при развитии нейрональной дегенерации подтверждаются многими авторами. Кроме того, в значительной части случаев ингибиторы кальпаинов предупреждают или замедляют развитие патологических процессов в нервной клетке. Однако принципиальным отличием данных работ является то, что акцент делается именно на гибель тела нейрона. Изменения, проходившие в отростках этих клеток, во внимание не принимались. Как бы там ни было, применение ингибиторов кальпаинов при лечении аксональной дегенерации может действительно оказаться тем фактором, который будет играть решающую роль.
Чем обусловлена аксональная дегенерация при амиотрофическом латеральном склерозе?
Причина или несколько причин аксональной дегенерации при БАС у мутантной линии мышей по супероксиддисмутазе не выяснена. Некоторые ученые рассматривают ее токсическое действие на клеточные элементы. Механизмы токсического влияния включают в себя агрегацию мутантных протеинов, поломку митохондриальных систем и токсическое действие глутамата. Все перечисленные факторы в отдельности или ассоциированно с факторами, вызывающими запуск апоптоза, приводят к характерным изменениям в клетке. Каждый из этих процессов достаточно хорошо изучен в отдельности и применительно к спинальным мотонейронам. Однако комплексный анализ с учетом особенностей процессов, протекающих в дистальных отделах аксонов, не проводился.
Дефекты аксонального транспорта
Процесс ухудшения работы быстрого антероградного аксонального транспорта у мутантных мышей по супероксиддисмутазе и у людей с БАС достаточно глубоко изучен. Заметное снижение скорости антероградного транспорта по аксонам с накоплением нейрофиламентов в проксимальной части нейрона описано у мутантных мышей и людей с БАС. При этом в спинном мозге признаки такого явления появляются в среднем за полгода до первых клинических проявлений заболевания. Замедление ретроградного транспорта также замечено в моторных нейронах мутантных мышей с 13-го дня эмбрионального развития. Такие явления, как изменение скорости транспорта веществ по отросткам нейрона, могут быть прямой причиной нарушения ее функции, а также следствием и симптомом такого нарушения. Выяснение истинной роли изменения скорости аксонального транспорта в аксональной дистрофии еще предстоит выяснить. Недавно были идентифицированы мутации, проявляющиеся в изменении комплекса белковых молекул с динеин/динактином, которые являются характерными именно для пораженных двигательных нейронов у мышей и людей. Нормализуя аксональный транспорт, достаточно часто удавалось пролонгировать жизнедеятельность клетки. Указанные данные свидетельствуют о том, что активность и состоятельность аксонального транспорта является еще одним критическим звеном в цепи патогенеза аксонального амиотрофического склероза.
Оксидативный стресс
При описании БАС и некоторых других нейродегенеративных процессов традиционно указывается на роль оксидативного повреждения как фактора, обусловливающего старение клетки и/или организма. Абсолютно очевидно, что в трупных тканях оксидативный процесс в отношении белков, липидов и ДНК при БАС выражен исключительно сильно. Однако какова роль перекисного окисления в формировании ранних стадий заболевания, на сегодня не выяснено. Аналогично выявлена роль оксидативного повреждения клеток у линии мутантных мышей. Абсолютно очевидно повышение интенсивности перекисного окисления в клетках спинного мозга после месячного периода развития симптомов болезни. Информация, касающаяся интенсивности оксидативного повреждения клеток в период, предшествующий появлению яркой симптоматики, является весьма скудной. Исходя из того, что абсолютное большинство клеточных элементов, белков, частей цитоскелета и др. попадают в дистальные части аксонов за счет аксонального транспорта, логично предположить, что именно дистальная часть отростка нейрона содержит наиболее «старые» клеточные единицы. Эти части клетки в большей мере подвергались травмирующему действию перекисного окисления, вследствие чего резервы и «запас прочности» у таких элементов снижены, а это значит, что дегенерация и дисфункция этих компонентов наиболее вероятна.
Несомненно то, что нейромышечные соединения могут быть уязвимы для повреждения окислительными процессами, активными у животных с мутацией по супероксиддисмутазе. До тех пор, пока у них не развивается дистрофия двигательных нейронов, значительных симптомов заболевания не наблюдается. И только после развития выраженной моторной аксонопатии происходит резкое ускорение процессов старения и появление мышечной атрофии. Миографические исследования показывают спонтанную активность пораженных мышц, что свидетельствует о начавшейся денервации. Среди миографических симптомов данного состояния встречаются фибрилляции и сложные комплексные разряды. Анализируя материалы гистологического исследования, можно сказать о начинающейся денервации дистальных отделов конечностей на втором месяце заболевания, и к концу первого полугодия формируется комбинация атрофии мышечных волокон и их гипертрофии. Кроме того, среди характерных признаков наблюдается группирование мышечных волокон, что указывает на хроническую моторную невропатию.
Предложена гипотеза о том, что клиническое и патофизиологическое сходство между разными группами мутантных мышей, отличавшихся только степенью выраженности экспрессии генов, кодирующих супероксиддисмутазу, заключается лишь в выраженности симптомов. Главный смысл состоит в том, что оксидативный стресс играет основную роль в аксональной дегенерации, наблюдающейся у животных данных групп. Интенсивность оксидативного стресса может быть достаточной для формирования или запуска процессов аксональной патологии. Например, увеличение размера митохондрий в нейромышечном соединении становится заметным на 25-й день развития патологического процесса в нервной ткани. Это указывает на значительное повреждение митохондриальной структуры. Оксидативное повреждение митохондрий нарушает динамику электронов, тем самым угнетая синтез АТФ, вплоть до его остановки. Это является еще одним фактором, замедляющим аксональный энергозависимый транспорт. Недостаточность энергетического снабжения транспортных систем нейрона приводит к остановке двустороннего обмена между телом и отростками нейрона. Замедление транспорта является нормальным проявлением старения нервных клеток. Как было описано, включается «порочный круг», связанный с увеличением оксидативного влияния на органеллы. АТФ также является жизненно важным компонентом для работы Na+/K+-АТФазы. Дисфункция этого фермента влечет за собой накопление натрия в аксоне. Повышение концентрации данного иона приводит к реверсу натрий-кальциевого транспорта с входом и накоплением кальция в аксоне. Как было описано выше, кальций является прямым активатором кальпаиновой системы, что резко ускоряет дегенеративные процессы в аксональных структурах. Помимо повреждения митохондрий, оксидативный стресс влияет на жизнедеятельность других клеточных компонентов, содержащихся в аксоне. Накопление гиперфосфорилированных нейрофиламентов в дистальных отделах аксонов — типичное явление для пациентов с БАС. Этот феномен является типичным для состояний, связанных с повышением активности Cdk5, встречающимся при оксидативном стрессе. В дополнение к огромному количеству эффектов, к которым приводит оксидативный стресс, свободные радикалы резко снижают эффективность выделения медиаторов из пресинапса моторных терминалей. Эксперименты, модулирующие аналогичные состояния за счет применения ботулина, имели стандартный и закономерный результат, заключающийся в повреждении дистальных отделов моторных аксонов, и даже приводили к гибели двигательных нейронов.
Влияние свободных радикалов на компоненты клеток невероятно многообразно. Оно проявляется в изменении свойств белковых и липидных молекул, что вызывает различные дегенеративные процессы. Перекисное окисление мутантной супероксиддисмутазы, как известно, приводит к прекращению функционирования этого фермента. Действие перекисного окисления на убиквитин-карбокси-терминальную гидролазу аналогично ее полной элиминации из клетки. Вследствие этого возникает так называемая грациллярная аксональная дистрофия. Обращает на себя внимание то, что нейрофиламенты исключительно чувствительны к оксидативному повреждению. Экспозиция очищенных нейрофиламентов в супероксиддисмутазе и пероксиде водорода вызывает дозозависимую агрегацию указанных белков. Агрегированные нейрофиламенты за счет действия любого из возможных факторов перекисного окисления, в частности аскорбата трихлористого железа, становятся более чувствительными к действию кальпаинов. Это, в свою очередь, приводит к заметному увеличению скорости дегенерации. В клетках нейробластомы человека высокие дозы аскорбата вызывают дегенерацию нейрофиламентов, имеющую дозозависимый эффект, что впоследствии приводит к гибели клетки. В отличие от нейрофиламентов тубулин и актин не подвергаются агрегации в такой модели перекисного окисления. Это означает, что нейрофиламенты имеют практически уникальную чувствительность к оксидативному стрессу. Такая их особенность может быть связана с наличием специфических локусов, сопряженных с лизином и пролином. Эти части белковой молекулы теоретически становятся мишенью для модификации свободными радикалами, что приводит к альтерации и изменению вторичной структуры белка. В модели цитоскелета свободные радикалы приводят к таким критическим изменениям белковых структур, которые вызывают его деструкцию. Данные состояния могут быть предупреждены или уменьшены введением витаминов Е и С.
Особенности проявления аксональной дегенерации указывают на исключительную важность оксидативных процессов в ее формировании. Так, денервация моторных соединений у больных мышей происходит по определенной закономерности. Первыми дегенерируют нейроны и их отростки, имеющие малый и средний уровень резервов. Медленные моторные единицы в меньшей степени подвержены дегенерации. Кроме того, в них быстрее происходит регенерация и восстановление отростков. Общая картина развития нейромышечных заболеваний отражает естественные процессы старения. Единственное отличие — скорость развития этих состояний. Интересным фактом является то, что наиболее уязвимы для оксидативного стресса клетки, в которых уровень окислительного фосфорилирования не слишком высок. Обычно именно в таких клетках количество элементов антиоксидантной системы гораздо ниже. Соответственно склонность к дегенеративным процессам нервной ткани может быть диагностирована по количеству и активности антиоксидантов в данном организме. Еще одним интересным наблюдением является то, что клетки, находящиеся в непосредственной близости к поврежденным элементам, увеличиваются в объеме. Также проявляется увеличенное количество мышечных элементов, компенсаторно иннервируемое ими. Предполагаемой причиной развития дисбаланса между процессами перекисного окисления и антиоксидантной системой может быть слишком быстрая экспрессия мутантного гена, кодирующего синтез супероксиддисмутазы, сдвигающее равновесие систем в сторону перекисного окисления и оксидативного стресса.
Многочисленные антиоксиданты тестировались при выполнении данного исследования. Некоторые из них проявили значительную антиоксидантную активность. Как результат удалось пролонгировать выживаемость мутантных мышей до двух раз относительно контрольной группы. При этом назначение антиоксидантов проводилось в период скрытого, бессимптомного течения заболевания. Тестирование активности антиоксидантных систем организма на пациентах, по результатам метаанализа, не принесло статистически значимого результата. Вследствие того, что измерение степени оксидативного стресса на сегодняшний день не представляется возможным, не удается установить причину отсутствия результата назначение антиоксидантной терапии пациентам. Так, неэффективность лечения может быть связана и с повышением степени окислительных реакций, и с недостаточной эффективностью предлагаемой терапии. Даже если оксидативный процесс играет одну из основных ролей в запуске цепочки реакций, приводящих к аксональной дегенерации, мы не можем с помощью медикаментов предотвратить этот процесс. Таким образом, для разработки эффективной методики лечения оксидативных повреждений при БАС нам необходимо еще более глубокое исследование цепочки патологических процессов, приводящих к этим изменениям.
Роль других клеток
В предшествующих разделах обсуждались явления в нейроне и его частях, которые могут играть важную роль в аксональной дегенерации. Несмотря на несомненно важную роль процессов, происходящих непосредственно в самой клетке, однозначным является то, что дегенеративные изменения при БАС не проходят изолированно только в нейронах. Реакции глиальных клеток также являются достаточно важным прогностическим критерием и признаком степени изменений, уже произошедших в ЦНС. Менее известны феномены, происходящие в клетках, находящихся в непосредственной близости от дистальных участков аксонов. К таким клеточным элементам можно отнести шванновские, мышечные и даже иммунные клетки.
Шванновские клетки являются наиболее крупными глиальными клетками, находящимися в ЦНС. Они имеют контакт с аксонами нервных клеток на огромной площади. Эти участки превосходят места контактов нейронов с астроцитами в несколько десятков раз. Не следует упускать из виду то, что эти клетки являются еще и значительными источниками трофической поддержки и физической защиты нейронов. Таким образом, можно считать, что именно шванновские клетки и нейроглиальные союзы являются наиболее подходящими объектами исследования для выяснения природы гибели нейронов. Мутантная супероксиддисмутаза обнаруживается в шванновских клетках мышей мутантной линии в первые дни постнатального периода. В терминальных шванновских клетках в острый период дегенерации нейронов наблюдается резкое повышение аксонального фактора семафорина-3А. Этот факт означает попытки шванновских клеток противостоять травмирующим факторам за счет синтеза ингибиторов перекисного окисления, проявляющих свойства стимуляторов регенерации. Таким образом, эти клетки глии на некоторое время задерживают начинающуюся дисфункцию нейромышечного соединения и замедляют развитие симптомов БАС.
При аналогичном анализе скелетной мускулатуры в миоцитах также обнаруживается высокий уровень мутантной супероксиддисмутазы, которая аналогичным образом действует на нейромышечное соединение. Некоторые авторы считают, что роль миоцитов заключается в запуске патологических реакций, направленных на дегенерацию нейромышечного соединения. Именно мышечная часть этого образования является причиной данной патологии. Однако достоверных данных, подтверждающих эту теорию, получено не было. Увеличение синтеза фактора роста и ветвления дендритов у пациентов с БАС и у животных мутантных линий вызвало создание гипотезы о возможной роли миоцитов в ингибировании регенеративных процессов дистальных отделов аксонов. В подтверждение этой гипотезы можно привести следующие факты: при ингибировании рядом ферментов указанных факторов роста дендритов увеличение продолжительности жизни мутантных мышей составило порядка 25 дней.
Последние несколько исследований были направлены на выяснение действительной роли миоцитов в формировании дегенерации нейронов у мутантных мышей. Отделение миоцитов из нейромышечного соединения не показало достоверных изменений продолжительности жизни животных мутантных линий. Кроме того, результатом данной работы было лишь незначительное снижение мутантных белков в исследуемых нейронах. Необходимо проведение дополнительных серий экспериментов, которые уточнят роль мышечных клеток в патологии двигательных нейронов. Кроме того, необходимо определить дальнейшие направления исследования для выяснения возможности использования компонентов миоцитов в лечении БАС.
Заключение
В результате масштабных исследований получены очевидные данные, касающиеся патогенеза нейрональной дегенерации. Неоспорим тот факт, что процессы аксональной дегенерации и изменения, происходящие в теле нейрона, наблюдаются независимо друг от друга. Поэтому чрезвычайно важно помнить, что мероприятия, направленные на поддержание жизнедеятельности тела нейрона, не повлияют на клинические проявления БАС и продолжительность жизни. С другой стороны, были выявлены критические зоны развивающейся патологии. С точки зрения авторов именно на них должна быть направлена терапия при дегенеративных состояниях нейронов. Механизм развития аксональной дегенерации до настоящего времени остается неизвестным. Однако с уверенностью можно сказать, что одной из важных фаз развития данных состояний является оксидативный стресс. Множество связанных с ним факторов могут внезапно спровоцировать резкое увеличение реакций перекисного окисления, что вызовет процессы ускоренного старения и, как следствие, дегенерации нервной ткани. В дополнение можно сказать, что роль шванновских клеток в развитии дегенеративных процессов нельзя недооценивать. Это связано именно со свойством данных клеток стимулировать регенеративные процессы в нейронах. Дальнейшие исследования, несомненно, дадут новые факты, которые позволят с иной стороны подойти к рассмотрению патогенеза дегенеративных заболеваний нервной ткани, в частности, к амиотрофическому латеральному склерозу.
Литература
1. Glass J.D. Wallerian degeneration as a window to peripheral neuropathy // J. Neurol. Sci. 2004; 220: 123-124.
2. Gould T.W., Buss R.R., Vinsant S., Prevette D. et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS // J. Neurosci. 2006; 26: 8774-8786.
3. Kostic V., Jackson-Lewis V., de Bilbao F., Dubois-Dauphin M., Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis // Science 1997; 277: 559-562.
4. Li M., Ona V.O., Guegan C., Chen M., Jackson-Lewis V. et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model // Science 2000; 288: 335-339.
5. Sagot Y., Dubois-Dauphin M., Tan S.A., de Bilbao F. et al. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease // J. Neurosci. 1995; 15: 7727-7733.
6. Klivenyi P., Ferrante R.J., Matthews R.T., Bogdanov M.B. et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis // Nat. Med. 1999; 5: 347-350.
7. Chiu A.Y., Zhai P., Dal Canto M.C., Peters T.M. et al. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis // Mol. Cell. Neurosci. 1995; 6: 349-362.
8. Kennel P.F., Finiels F., Revah F., Mallet J. Neuromuscular function impairment is not caused by motor neuron loss in PALS mice: an electromyographic study // Neuroreport. 1996; 7: 1427-1431.
9. Frey D., Schneider C., Xu L., Borg J., Spooren W., Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases // J. Neurosci. 2000; 20: 2534-2542.
10. Fischer L.R., Culver D.G., Tennant P., Davis A.A. et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man // Exp. Neurol. 2004; 185: 232-240.
11. Pun S., Santos A.F., Saxena S., Xu L., Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF // Nat. Neurosci. 2006; 9: 408-419.
12. Schaefer A.M., Sanes J.R., Lichtman J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis // J. Comp. Neurol. 2005; 490: 209-219.
13. Bommel H., Xie G., Rossoll W., Wiese S., Jablonka S., Boehm T., Sendtner M. Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease // J. Cell. Biol. 2002; 159: 563-569.
14. Martin N., Jaubert J., Gounon P., Salido E., Haase G., Szatanik M., Guenet J.L. A missense mutation in Tbce causes progressive motor neuronopathy in mice // Nat. Genet. 2002; 32: 443-447.
15. Holtmann B., Zielasek J., Tokya K.V., Sendtner M. Comparative analysis of motoneuron loss and functional deficits in PMN mice: implications for human motoneuron disease // J. Neurol. Sci. 1999; 169: 140-147.
16. Sendtner M., Schmalbruch H., Stockli K.A. et al. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy // Nature 1992; 358: 502-504.
17. Cifuentes-Dias C., Nicole S., Velasco M.E. et al. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model // Hum. Mol. Genet. 2002; 11: 1439-1447.
18. Hansen S., Ballantyne J.P. A quantitative electrophysiological study of motor neuron disease // J. Neurol. Neurosurg. Psychiatry 1978; 41: 773-783.
19. Bradley W.G., Good P., Rasool C.G., Adelman L.S. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis // Ann. Neurol. 1983; 14: 267-277.
20. Shefner J.M. Motor unit number estimation in human neurological diseases and animal models // Clin. Neurophysiol. 2001; 112: 955-964.
21. Armon C., Brandstater M.E. Motor unit number estimate-based rates of progression of ALS predict patient survival // Muscle Nerve 1999; 22: 1571-1575.
22. Yuen E.C., Olney R.K. Longitudinal study of fiber density and motor unit number estimate in patients with amyotrophic lateral sclerosis // Neurology 1997; 49: 573-578.
23. Shefner J.M., Cudkowicz M., Brown R.H. Jr. Motor unit number estimation predicts disease onset and survival in a transgenic mouse model of amyotrophic lateral sclerosis // Muscle Nerve 2006; 34: 603-607.
24.Aggarwal A., Nicholson G. Detection of pre-clinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation // J. Neurol. Neurosurg. Psychiatry 2002; 73: 199-201.
25. Kanai K., Kuwabara S., Misawa S., Tamura N. et al. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage // Brain 2006; 129: 953-962.
26.Vucic S., Kiernan M.C. Axonal excitability properties in amyotrophic lateral sclerosis // Clin. Neurophysiol. 2006; 117: 1458-1466.
27. Nakata M., Kuwabara S., Kanai K., Misawa S. et al. Distal excitability changes in motor axons in amyotrophic lateral sclerosis // Clin. Neurophysiol. 2006; 117: 1444-1448.
28. Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics // Nat. Rev. Neurosci. 2006; 7: 710-723.
29. Heads T., Pollock M., Robertson A., Sutherland W.H.F., Allpress S. Sensory nerve pathology in amyotrophic lateral sclerosis // Acta Neuropathol. 1991; 82: 316-320.
30. Kawamura Y., Dyck P.J., Shimono M., Okazaki H., Tateishi J., Doi H. Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis // J. Neuropathol. Exp. Neurol. 1981; 40: 667-675.
31. Shefner J.M., Tyler R., Krarup C. Abnormalities in the sensory action potential in patients with amyotrophic lateral sclerosis // Muscle Nerve 1991; 14: 1242-1246.
32. Theys P.A., Peelers E., Robberecht W. Evolution of motor and sensory deficits in amyotrophic lateral sclerosis estimated by neurophysiological techniques // J. Neurol. 1999; 246: 438-442.
33. Fischer L.R., Culver D., Davis A.A., Tennant P. et al. The WldS gene modestly prolongs survival in the SOD1-G93A fALS mouse // Neurobiol. Dis. 2005; 19: 293-300.
34. Anagnostou E., Miller S.P., Guiot M.C., Karpati G. et al. Type I spinal muscular atrophy can mimic sensory-motor axonal neuropathy // J. Child Neurol. 2005; 20: 147-150.
35. Rudnik-Schoneborn S., Goebel H.H., Schlote W. et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy // Neurology 2003; 60: 983-987.
36. Jablonka S., Karle K., Sandner B., Andreassi C., von Au K., Sendtner M. Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy // Human Mol. Genet. 2006; 15: 511-518.
37. Finn J.T., Weil M., Archer F., Siman R., Srinivasan A., Raff M.C. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases // J. Neurosci. 2000; 20: 1333-1341.
38. Sagot Y., Vejsada R., Kato A. Clinical and molecular aspects of motoneurone diseases: animal models, neurotrophic factors and Bcl-2 oncoprotein // Trends Pharmacol. Sci. 1997; 18: 330-337.
39. Lunn E.R., Perry V.H., Brown M.C., Rosen H., Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve // Eur. J. Neurosci. 1989; 1: 27-33.
40. Conforti L., Tarlton A., Mack T.G.A., Mi W. et al. A Ufd2/D4Colele chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wld s ) mouse // Proc. Natl. Acad. Sci. USA 2000; 97: 11377-11382.
41. Wang M.S., Davis A.A., Culver D.G., Glass J.D. WldS mice are resistant to paclitaxel (taxol) neuropathy // Ann. Neurol. 2002; 52: 442-447.
42. Wang M.S., Fang G., Culver D.G., Davis A.A., Rich M.M., Glass J.D. The Wld s protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy // Ann. Neurol. 2001; 50: 773-779.
43. Wang M.S., Wu Y., Culver D.G., Glass J.D. The gene for slow Wallerian degeneration (Wld s ) is also protective against vincristine neuropathy // Neurobiol. Dis. 2001; 8: 155-161.
44.Ferri A., Sanes J.R., Coleman M.P., Cunningham J.M., Kato A.C. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease // Curr. Biol. 2003; 13: 669-673.
45. Samsam M., Mi W., Wessig C., Zielasek J. et al. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy // J. Neurosci. 2003; 23: 2833-2839.
46. Deckwerth T.L., Johnson J., Eugene M. Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) // Dev. Biol. 1994; 165: 63-72.
47. Adalbert R., Nogradi A., Szabo A., Coleman M.P. The slow Wallerian degeneration gene in vivo protects motor axons but not their cell bodies after avulsion and neonatal axotomy // Eur. J. Neurosci. 2006; 24: 2163-2168.
48. Coleman M. Axon degeneration mechanisms: commonality amid diversity // Nat. Rev. Neurosci. 2005; 6: 889-898.
49. Raff M.C., Whitmore A.V., Finn J.T. Axonal self-destruction and neurodegeneration // Science 2002; 296: 868-871.
50. Coleman M.P., Perry V.H. Axon pathology in neurological disease: a neglected therapeutic target // Trends Neurosci. 2002; 25: 532-537.
51. Vande Velde C., Garcia M.L., Yin X., Trapp B.D., Cleveland D.W. The neuroprotective factor Wlds does not attenuate mutant SODl-mediated motor neuron disease // Neuromolecular Med. 2004; 5: 193-203.
52. Crawford T.O., Hsieh S.T., Schryer B.L., Glass J.D. Prolonged axonal survival in transected nerves of C57BL/Ola mice is independent of age // J. Neurocytol. 1995; 24: 333-340.
53. Gillingwater T.H., Thomson D., Mack T.G.A. et al. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld s mutant and Ube4b/Nmnat transgenic mice // J. Physiol. 2002; 543: 739-755.
54.Araki T., Sasaki Y., Millbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration // Science 2004; 305: 1010-1013.
55. Wang J., Zhai Q., Chen Y., Lin E., Gu W., McBurney M.W., He Z. A local mechanism mediates NAD-dependent protection of axon degeneration // J. Cell. Biol. 2005; 170: 349-355.
56. Conforti L., Fang G., Beirowski B., Wang M.S. et al. NAD(+) and axon degeneration revisited: Nmnatl cannot substitute for Wld(S) to delay Wallerian degeneration // Cell Death Differ. 2007; 14: 116-127.
57. Bartus R., Elliott P., Hayward N. Calpain as a novel target for treating acute neurodegenerative disorders // Neurol. Res. 1995; 17: 249-258.
58. Wang M.S., Wu Y., Culver D., Glass J.D. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincris-tine neuropathy // J. Neuropathol. Exp. Neurol. 2000; 59: 599-606.
59. Wang M.S., Davis A.A., Culver D.G., Wang Q., Powers J.C., Glass J.D. Calpain inhibition protects against Taxol-induced sensory neuropathy // Brain 2004; 127: 671-679.
60. O’Hanlon G.M., Humphreys P.D., Goldman R.S. et al. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury // Brain 2003; 126: 2497-2509.
61. Adamec E., Mohan P., Vonsattel J.P., Nixon R.A. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2 // Acta Neuropathol. 2002; 104: 92-104.
62. Nixon R.A., Saito K.I., Grynspan F., Griffin W.R. et al. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease // Ann. NY Acad. Sci. 1994; 747: 77-91.
63. Kieran D., Greensmith L. Inhibition of cal-pains, by treatment with leupeptin, improves motoneuron survival and muscle function in models of motoneuron degeneration // Neuroscience 2004; 125: 427-439.
64. Li J., Nixon R.A., Messer A., Berman S., Bursztajn S. Altered gene expression for calpain/calpastatin system in motor neuron degeneration (Mnd) mutant mouse brain and spinal cord // Brain Res. Mol. Brain Res. 1998; 53: 174-186.
65. Brown J., Robert H. Superoxide dismutase in familial amyotrophic lateral sclerosis: models for gain of function // Curr. Opin. Neurobiol. 1995; 5: 841-846.
66. Cleveland D.W., Rothstein J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS // Nat. Rev. Neurosci. 2001; 2: 806-819.
67. Boillee S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors // Neuron 2006; 52: 39-59.
68. Williamson T.L., Cleveland D.W. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons // Nat. Neurosci. 1999; 2: 50-56.
69. Zhang B., Tu P.H., Abtahian F., Trojanowski J.Q., Lee V.M.Y. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation // J. Cell. Biol. 1997; 139: 1307-1315.
70. Warita H., Itoyama Y., Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene // Brain Res. 1999; 819: 120-131.
71. Sasaki S., Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis // Neurology 1996; 47: 535-540.
72. Murakami T., Nagano I., Hayashi T., Manabe Y. et al. Impaired retrograde axonal transport of adenovirus-mediated E. coli LacZ gene in the mice carrying mutant SOD1 gene // Neurosci. Lett. 2001; 308: 149-152.
73. Kieran D., Hafezparast M., Bohnert S. et al. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice // J. Cell. Biol. 2005; 169: 561-567.
74. Hafezparast M., Klocke R., Ruhrberg C. et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport // Science 2003; 300: 808-812.
75. LaMonte B., Wallace K.E., Holloway B.A. et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive deterioration // Neuron 2002; 34: 715-727.
76. Puls I., Jonnakuty C., LaMonte B., Holzbaur E.L.F. et al. Mutant dynactin in motor neuron disease // Nat. Genet. 2003; 33: 455-456.
77. Balaban R.S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging // Cell 2005; 120: 483-495.
78. Barber S.C., Mead R.J., Shaw P.J. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target // Biochim. Biophys. Acta 2006; 1762: 1051-1067.
79. Hall E.D., Andrus P.K., Oostveen J.A., Fleck T.J., Gurney M.E. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS // J. Neurosci. Res. 1998; 53: 66-77.
80. Shefner J.M., Reaume A.G., Flood D.G. et al. Mice lacking cytosolic copper/zinc superoxide dismutase display a distinctive motor axonopathy // Neurology 1999; 53: 1239-1246.
81. Muller P.L., Song W., Liu Y. et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy // Free Radic. Biol. Med. 2006; 40: 1993-2004.
82. Flood D.G., Reaume A.G., Gruner J.A., Hoffman E.K. et al. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions // Am. J. Pathol. 1999; 155: 663-672.
83. Echtay K.S., Roussel D., St-Pierre J., Jekabsons M.B. et al. Superoxide activates mitochondrial uncoupling proteins // Nature 2002; 415: 96-99.
84. Fernandez H.L., Hodges-Savola C.A. Axoplasmic transport of calcitonin gene-related peptide in rat peripheral nerve as a function.of age // Neurochem. Res. 1994; 19: 1369-1377.
85.Frolkis V.V., Tanin S.A., Gorban Y.N. Age-related changes in axonal transport // Exp. Gerontol. 1997; 32: 441-450.
86. Shea T.B., Zheng Y.L., Ortiz D., Pant H.C. Cyclin-dependent kinase 5 increases perikaryal neurofilament phosphorylation and inhibits neurofilament axonal transport in response to oxidative stress // J. Neurosci. Res. 2004; 76: 795-800.
87. Giniatullin A.R., Darios F., Shakirzyanova A., Davletov B., Giniatullin R. SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release // J. Neurochem. 2006; 98: 1789-1797.
88. Berliocchi L., Fava E., Leist M., Horvat V., Dinsdale D., Read D., Nicotera P. Botulinum neurotoxin C initiates two different programs for neurite degeneration and neuronal apoptosis // J. Cell Biol. 2005; 168: 607-618.
89. Hodgson E.K., Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme // Biochemistry 1975; 14: 5294-5299.
90. Poon H.P., Hensley K., Thongboonkerd V., Merchant M.L. et al. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice — a model of familial amyotrophic lateral sclerosis // Free Radic. Biol. Med. 2005; 39: 453-462.
91. Saigoh K., Wang Y.L., Suh J.G., Yamanishi T., Sakai Y. et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice // Nat. Genet. 1999; 23: 47-51.
92. Kim N.H., Jeong M.S., Choi S.Y., Hoon Kang J. Oxidative modification of neurofilament-L by the Cu,Zn-superoxide dismutase and hydrogen peroxide system // Biochimie 2004; 86: 553-559.
93. Troncoso J.C., Costello A.C., Kim J.H., Johnson G.V. Metal-catalyzed oxidation of bovine neurofilaments in vitro // Free Radic. Biol. Med. 1995; 18: 891-899.
94. Cookson M.R., Thatcher N.M., Ince P.G., Shaw P.J. Selective loss of neurofilament proteins after exposure of differentiated human IMR-32 neuroblastoma cells to oxidative stress // Brain Res. 1996; 738: 162-166.
95. Gelinas S., Chapados C., Beauregard M., Gosselin I., Martinoli M.G. Effect of oxidative stress on stability and structure of neurofilament proteins // Biochem. Cell Biol. 2000; 78: 667-674.
96. Counterman A.E., D’Onofrio T.G., Andrews A.M., Weiss P.S. A physical model of axonal damage due to oxidative stress // Proc. Natl. Acad. Sci. USA 2005; 103: 5262-5266.
97. Gordon T., Hegedus J., Tarn S.L. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease // Neurol. Res. 2004; 26: 174-185.
98. Luff A.R. Age-associated changes in the innervation of muscle fibers and changes in the mechanical properties of motor units // Ann. NY Acad. Sci. 1998; 854: 92-101.
99. Hollander J., Fiebig R., Gore M., Bejma J., Ookawara T., Ohno H., Ji L.L. Superoxide dismutase gene expression in skeletal muscle: fiber-specific adaptation to endurance training // Am. J. Physiol. 1999; 277: R856-R862.
100. Crow J.P., Calingasan N.Y., Chen J., Hill J.L., Beal M.F. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice // Ann. Neurol. 2005; 58: 258-265.
101. Liu R., Li B., Flanagan S.W., Oberley L.W., Gozal D., Qiu M. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival // J. Neurochem. 2002; 80: 488-500.
102. Orrell R.W., Lane R.J., Ross M. Antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease // Cochrane Database Syst. Rev. 2005; 1: CD002829.
103. Lino M.M., Schneider C., Caroni P. Accumulation of SOD1 mutants in postnatal moto-neurons does not cause motoneuron pathology or motoneuron disease // J. Neurosci. 2002; 22: 4825-4832.
104. Pramatarova A., Laganiere J., Roussel J., Brisebois K., Rouleau G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment // J. Neurosci. 2001; 21: 3369-3374.
105. Boillee S., Yamanaka K., Lobsiger C.S., Copeland N.G. et al. Onset and progression in inherited ALS determined by motor neurons and microglia // Science 2006; 312: 1389-1392.
106. Clement A., Nguyen M., Roberts E., Garcia M. et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice // Science 2003; 302: 113-117.
107. Hall E.D., Oostveen J.A., Gurney M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS // Glia 1998; 23: 249-256.
108. Howland D.S., Liu J., She Y. et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) // Proc. Natl. Acad. Sci. USA 2002; 99: 1604-1609.
109. Di Giorgio F.P., Carrasco M.A., Siao M.C., Maniatis T., Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model // Nat. Neurosci. 2007; 10: 608-614.
110. Nagai M., Re D.B., Nagata T., Chalazonitis A. et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons // Nat. Neurosci. 2007; 10: 615-622.
111. De Winter F., Vo T., Stam F.J., Wisman L.A. et al. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease // Mol. Cell. Neurosci. 2006; 32: 102-117.
112. Turner B.J., Lopes E.C., Cheema S.S. Neuromuscular accumulation of mutant superoxide dismutase 1 aggregates in a transgenic mouse model of familial amyotrophic lateral sclerosis // Neurosci. Lett. 2003; 350: 132-136.
113. Taylor A.R., Gifondorwa D.J., Newbern J.M, Robin-son M.B. et al. Astrocyte and muscle-derived secreted factors differentially regulate motoneuron survival // J. Neurosci. 2007; 27: 634-644.
114. Dupuis L., Gonzalez de Aguilar J.L., di Scala F., Rene F., de Tapia M. et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis // Neurobiol. Dis. 2002; 10: 358-365.
115. Jokic N., Gonzalez de Aguilar J.L., Dimou L. et al. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model // EMBO Rep. 2006; 7: 1162-1167.
116. Miller T.M., Kim S.H., Yamanaka K. et al. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis // Proc. Natl. Acad. Sci. USA 2006; 103: 19546-19551.
117. Holzbaur E.L., Howland D.S., Weber N. et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis // Neurobiol. Dis. 2006; 23: 697-707.
118. Dobrowolny G., Giacinti C., Pelosi L., Nicoletti C. et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model // J. Cell. Biol. 2005; 168: 193-199.
119. Cifuentes-Diaz C., Frugier T., Tiziano F.D., Lacene E. et al. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy // J. Cell. Biol. 2001; 152: 1107-1114.
120. Rosen D., Siddique T., Patterson D., Figlewicz D., Sapp P. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis // Nature 1993; 362: 59-62.
121. Ranta S., Zhang Y., Ross B., Lonka L., Takkunen E. et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8 // Nat. Genet. 1999; 23: 233-236.
122. Lefebvre S., Burglen L., Reboullet S., Clermont O. et al. Identification and characterization of a spinal muscular atrophy-determining gene // Cell 1995; 80: 155-165.
123. McWhorter M.L., Monani U.R., Burghes A.H., Beat-tie C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding // J. Cell. Biol. 2003; 162: 919-931.
124. Rich M.M., Wang X., Cope T.C., Pinter M.J. Reduced neuromuscular quantal content with normal synaptic release time course and depression in canine motor neuron disease // J. Neurophysiol. 2002; 88: 3305-3314.
125. La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy // Nature 1991; 352: 77-79.
126. Katsuno M., Adachi H., Minamiyama M., Waza M. et al. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration // J. Neurosci. 2006; 26: 12106-12117.
127. Chambers D.M., Peters J., Abbott C.M. The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor lalpha, encoded by the Eefla2 gene // Proc. Natl. Acad. Sci. USA 1998; 95: 4463-4468.
128. Newbery H.J., Gillingwater T.H., Dharmasaroja P. et al. Progressive loss of motor neuron function in wasted mice: effects of a spontaneous null mutation in the gene for the eEF1 A2 translation factor // J. Neuropathol. Exp. Neurol. 2005; 64: 295-303.
129. Schmitt-John T., Drepper C., Mussmann A., Hahn P. et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse // Nat. Genet. 2005; 37: 1213-1215.
130. Blondet B., Carpentier G., Ait-Ikhlef A., Murawsky M., Rieger F. Motoneuron morphological alterations before and after the onset of the disease in the wobbler mouse // Brain Res. 2002; 930: 53-57.
131. Oosthuyse B., Moons L., Storkebaum E., Beck H. et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration // Nat. Genet. 2001; 28: 131-138.
132. Miura H., Oda K., Endo C., Yamazaki K., Shibasaki H., Kikuchi T. Progressive degeneration of motor nerve terminals in GAD mutant mouse with hereditary sensory axonopathy // Neuropathol. Appl. Neurobiol. 1993; 19: 41-51.
133. Liedtke W., Leman E.E., Fyffe R.E., Raine C.S., Schubart U.K. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems // Am. J. Pathol. 2002; 160: 469-480.
134. Dewil M., dela Cruz V.F., Van Den Bosch L., Robberecht W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1G93A -induced motor neuron death // Neurobiol. Dis. 2007; 26: 332-341.
135. Rouaux C., Panteleeva I., Rene F., Gonzalez de Aguilar J.L. et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model // J. Neurosci. 2007; 27: 5535-5545.
ЭЛИ-Н-Тест-12
![]()
Алексеев Андрей Викторович
Врач клинической лабораторной диагностики,
Срок исполнения
(рабочие дни):
до 10 дн.
* Взятие биоматериала оплачивается отдельно
Анализ крови ЭЛИ-Н-Тест
 Иммунологический анализ крови «ЭЛИ-Н-Тест», включающий 12 исследований для определения уровня содержания аутоиммунных антител к антигенным компонентам клеток человеческого организма, проводится для оценивания состояния нейроглий, нейронов и нервных волокон, прогнозирования риска возникновения патологий нервной системы, их ранней диагностики и мониторинга эффективности проводимых лечебных мероприятий.
Иммунологический анализ крови «ЭЛИ-Н-Тест», включающий 12 исследований для определения уровня содержания аутоиммунных антител к антигенным компонентам клеток человеческого организма, проводится для оценивания состояния нейроглий, нейронов и нервных волокон, прогнозирования риска возникновения патологий нервной системы, их ранней диагностики и мониторинга эффективности проводимых лечебных мероприятий.
Нервная система человека является функциональной совокупностью взаимосвязанных нейронов, которые обеспечивают связь организма с внешней средой и регуляцию деятельности всех органов и систем. Ее структуру составляют центральный (в состав которого входит головной и мозг) и периферический (состоящий из нервных волокон, обеспечивающих двигательную активность человеческого тела) отделы. Нарушение их деятельности негативно сказывается на работе всего организма. Для диагностирования патологических процессов, протекающих в нервной системе, на ранних стадиях развития практикующие специалисты используют «ЭЛИ-Н-Тест-12», который представляет собой лабораторное исследование, позволяющее определить сывороточную концентрацию аутоиммунных антител к антигенам:
- NF-200.
- GFAP (специфического протеина филаментов астроцитов).
- S100 (регулятора клеточных функций).
- ОБМ (специфического белка миелиновых оболочек аксонов).
- Вольтаж-зависимого Са-канала.
- Холинорецепторов.
- Глутаматных рецепторов.
- ГАМК-рецепторов.
- Дофаминовых рецепторов.
- Серотониновых рецепторов.
- Опиатных mu-рецепторов.
- В-Эндорфина.
Суть анализа крови ЭЛИ-Н-Тест
Основное предназначение нервной системы человека – выполнение фундаментальных функций. Она способна контролировать деятельность абсолютно всех систем и органов. Тесно взаимодействуя с эндокринной системой, нервная система не только создаёт синхронное регулирование всех систем в организме, но и оказывает ответное воздействие на адаптацию к условиям внешней, внутренней среды.
Кроме того, нервной системе свойственна интегративная функция, базирующаяся на коммутации регуляторных систем, двигательной активности и чувствительности. Её систематическая диагностика является обязательным критерием полноценной жизни.
Комплексная лабораторная диагностика 12 антигенов ЭЛИ-Н направлена на оценку деятельности нервной системы двенадцатью антителами-маркерами с целью обнаружения задолго до возникновения первых признаков болезни различных патологий, а также предрасположенности к ним.
Тест оценки нервной системы показан пациентам с затруднённой постановкой диагноза, в случаях, когда требуется оценить риск возникновения определённых заболеваний (например, у людей с наследственной предрасположенностью к тем или иным патологиям), либо когда необходимо проанализировать динамику терапии и определить её эффективность.
Специфика антител ЭЛИ-Н-Тест
 NF-200 – специфический белок нейритов (аксонов). В процессе дегенерации покрытых глиальной оболочкой отростков нервных клеток, обеспечивающих передачу нервных импульсов, лежит увеличение антител к определённому белку.
NF-200 – специфический белок нейритов (аксонов). В процессе дегенерации покрытых глиальной оболочкой отростков нервных клеток, обеспечивающих передачу нервных импульсов, лежит увеличение антител к определённому белку.
GFAP – белок внутриклеточных нитевидных образований астроцитов. Повышение титра антител к нему говорит об аномальной активной фазе глиоза (разрастании клеток астроглии).
S100 – кальций-зависимый регуляторный белок, принимающий участие в большинстве клеточных процессов, таких как регуляция программирования клеточной гибели, трофический показатель СТ-ергических нейронов и пр. Увеличение антител к нему происходит преимущественно посредством видоизменений эмоционального самочувствия. Нередко основанием роста антител к специфическому белку S100 согласно концепции феномена молекулярной мимикрии является папилломавирус.
ОБМ – одна из разновидностей белка оболочки мякотного нервного волокна аксонов. Повышение антител к данному белку свидетельствует о наличии патологий в нервных волокнах, в т. ч. демиелинизирующих заболеваний.
Вольтажзависимый кальциевый канал входит в группу специфических антигенов. Повышение титра антител к нему часто является предпосылкой развития миастенического синдрома Ламберта-Итона (МСЛИ), латерального амиотрофического склероза (ЛАС), мозжечковой атаксии.
Аномальный рост концентрации антител к соединяющему домену дофаминовых, серотониновых, глутаматных нейромедиаторов, а также рецепторов гамма-аминомасляной кислоты (ГАМК) с лигандом встречается довольно часто, и подтверждает наличие изменений в конкретных механизмах нейронов. Данный тест способствует проведению недифференцированного анализа содержания в сыворотке крови антител к А- и В-ГАМК, дофаминовым Д1 и Д5, глутаматным AMPA, NMDA, серотониновым 5-Н1 и 5-Н7, н-холинорецепторам.
Излишек антител к beta2-гликопротеину, ДНК подтверждает нехарактерную иммуноактивацию, преимущественно совокупную с нестандартным воспалительно-инфекционным процессом.
Что показывает тест?
С помощью ЭЛИ-Н-Тест-12 врач может не только составить полную картину работы нервной системы, но и подобрать / скорректировать индивидуальный план лечения, выявить пациентов группы риска на фоне наследственной предрасположенности к таким заболеваниям как деменция, болезнь Альцгеймера, инсульт.
В ходе лабораторного теста на состояние ЦНС сыворотка крови исследуется на наличие в ней маркеров-антител к двенадцати антигенам ЦНС. Выявление маркеров свидетельствует о возможных патологических процессах в организме. К примеру, переизбыток антител к NF-200 является сигналом гибели нервных волокон, а их недостаток к beta2-гликопротеину, ДНК – признаком развития воспалительного процесса, вызванного инфекцией.
Когда делают анализ
Показаниями для проведения иммунологического исследования крови ЭЛИ-Н-Тест являются:
- скрининг структурно-функционального состояния нервной системы пациента;
- прогнозирование возможности возникновения тяжелых нервных расстройств у лиц с генетической предрасположенностью;
- постановка точного диагноза при общетерапевтических заболеваниях;
- объективное оценивание успешности назначенного лечения.
Подготовка и метод исследования

Анализ проводят с помощью метода ИФА, основанного на иммунной реакции. Для его выполнения используют образец венозной крови, который отбирают из локтевой вены в лабораторных условиях в утренние часы, натощак. Накануне процедуры пациенту необходимо воздержаться от:
- пищевой нагрузки;
- чрезмерного физического и психоэмоционального напряжения;
- употребления алкогольных напитков;
- применения лекарственных препаратов, которые искажают итоговые данные исследования.
Расшифровка анализа
В норме параметры интервалов для маркерных аутоиммунных антител варьируют в пределах от -15 до 10%.
На основании итоговых данных тестов квалифицированный специалист проводит оценивание аутомаркеров – увеличение их концентрации указывает на изменение функционирования взаимосвязанных нервных структур пациента.
Расшифровка результатов выполняется после диагностики уровня 12 специфических антител, отражающих функциональную деятельность нервной системы. Состояние иммунной системы оценивается на основании анализа отклонений от референсных показателей. С учётом расхождения полученных результатов исследования с нормами подтверждается / опровергается тот или иной диагноз.
Изменения иммунореактивности в сторону повышения концентрации специфических аутоантител более 20 % в сравнении с индивидуальными средними нормами являются показателем потенциального индикатора патологических процессов в определённом органе.
Рост титра протеида аксонов характерный для заболеваний, при которых нервным импульсам свойственно проникать к иннервируемым органам.
Нарушение нормативных значений миелина в нейронах свойственно патологическим изменениям, отвечающим за поток нервных импульсов.
 Увеличение концентрации GFAP может быть показателем нейроэктодермального злокачественного образования головного мозга, образующегося из астроцитов.
Увеличение концентрации GFAP может быть показателем нейроэктодермального злокачественного образования головного мозга, образующегося из астроцитов.
Рост титра антител, связывающих домен с лигандом, характерный для состояний, связанных с неправильным обменом серотонина, холина, глутамата, гамма-аминомасляной кислоты, дофамина.
Прохождение исследования даёт возможность вовремя обнаружить изоморфные и функциональные патологии нервной системы. Диагностика нужна не столь для профилактических мер и подбора методов лечения, сколько для постановки окончательного диагноза.
Результаты исследования ЭЛИ-Н-Тест-12 дают возможность специалистам диагностического центра разработать профилактическую программу заболеваний нервной системы либо подобрать схему индивидуального лечения. Пройти обследование у врача можно по предварительной записи.